Эта статья опубликована под лицензией Creative Commons и не автором статьи. Поэтому если вы найдете какие-либо неточности, вы можете исправить их, обновив статью.

Отбор полиморфных локусов для анализа ассоциаций при генетико-эпидемиологических исследованиях



Ирина Васильевна Пономаренко
Опубликована Янв. 1, 2018
Последнее обновление статьи Сен. 19, 2022
Эта статья опубликована под лицензией
")



Аннотация
Изучение роли наследственных факторов в формировании многофакторных признаков является актуальным, а применительно к мультифакториальным заболеваниям эти исследования имеют важное значение для медицины. При планировании генетико-эпидемиологических исследований мультифакториальных признаков (заболеваний) важное значение имеет отбор полиморфных локусов для поиска ассоциаций с исследуемым фенотипом (заболеванием). Цель исследования. Провести систематический анализ данных, имеющихся в современной литературе, о подходах к отбору полиморфных локусов при проведении ассоциативных исследований. Материалы и методы. В обзор включены современные данные зарубежных и отечественных статей, найденные в Pubmed по данной теме. Результаты. Согласно современным представлениям при отборе полиморфных локусов генов-кандидатов для изучения ассоциаций с мультифакториальным признаком (заболеванием) следует учитывать следующие критерии: 1) наличие ассоциации с исследуемым признаком по результатам ранее проведенных полногеномных (GWAS) и/или ассоциативных (в том числе репликативных) исследований; 2) наличие ассоциации с фенотипами, имеющими с исследуемым признаком, общие биологические пути; 3) регуляторный потенциал (regSNP); 4) влияние на экспрессию генов (eSNP); 5) связь с несинонимическими заменами (nsSNP); 6)tagger SNP (tagSNP) 7) частота полиморфизма не менее 5% 8) функциональные эффекты (regSNP, eSNP, nsSNP) SNPs, находящихся в неравновесии по сцеплению (r2≥0.8) с отобранными для ассоциативного анализа полиморфизмами. В работе приведена характеристика современных мировых баз данных по функциональной геномике и биоинформатических методов анализа, используемых для in silico анализа регуляторного и eQTL значения SNPs, оценки их блочной структуры (SIFT, PolyPhen-2, HaploReg, rSNPs MAPPER, RegulomeDB, rSNPBase, SNP FuncPred, Blood eQTL browser, GTExportal, HaploView, LD TAG SNP Selection). Заключение. При отборе полиморфных локусов для ассоциативных исследований следует учитывать их ассоциации с исследуемым признаком по данным ранее проведенных исследований, регуляторный потенциал и влияние на экспрессию генов, nsSNP и tagSNP, популяционную частоту не менее 5%, функциональные эффекты сильно сцепленных с ними SNPs.
Ключевые слова
Ассоциации, регуляторный потенциал, eSNP, nsSNP, полиморфизм, tagSNP
Введение.
Одним из ключевых моментов любого генетико-эпидемиологического исследования, направленного на поиск ассоциаций полиморфных локусов генов-кандидатов с формированием многофакторного признака (как нормального, например, рост, вес, возраст менархе и др., так патологического, т.е. мультифакториального заболевания) является выбор полиморфных локусов определенных генов-кандидатов для исследования. Именно от того насколько «качественно» исследователь, планирующий свою работу, отберет полиморфные локусы для анализа ассоциаций будет напрямую зависеть «успех» его научного поиска. Если при отборе генов-кандидатов для исследования минимальным требованием для его включения в исследование является вовлеченность продукта данного гена в механизмы развития исследуемого многофакторного признака (этиопатогенез мультифакториального заболевания) (эту информацию можно получить в многочисленных руководствах по молекулярным механизмам развития различных заболеваний, а также в современных онлайн базах данных - KEGGPATHWAY (www.genome.jp/kegg/pathway.html), ReactomePathway (www.reactome.org), GeneOntology (http://geneontology.org), PharmGKB (www.pharmgkb.org), GeneMANIA (http://genemania.org) и др.), то при отборе полиморфных локусов, планируемых для исследования в данных генах-кандидатах, следует учитывать не только локализацию данного полиморфизма в/около рассматриваемого гена-кандидата и его ассоциации с данным фенотипом (заболеванием), полученные в ранее проведенных исследованиях в различных популяциях, но и его потенциальные фенотипические эффекты, основанные на связи данного полиморфизма с эпигенетическими изменениями и экспрессией генов. Именно эти доказанные (а не гипотетические) фенотипические эффекты «значимых» полиморфных локусов могут являться медико-биологической основой выявленных ассоциаций и должны учитываться при планировании любого генетико-эпидемиологического исследования. К настоящему времени в современной литературе накоплен значительный фактический материал, являющийся результатом работы достаточно большого числа международных консорциумов в области изучения эпигенетики, экспрессии генов человека – ENCODE (The Encyclopedia of DNA Elements) Consortium (https://www.encodeproject.org/), NIH Roadmap Epigenomics Mapping Consortium (http://www.roadmapepigenomics.org/), Consortium Genotype-Tissue Expression (http://www.gtexportal.org/), что диктует необходимость некоторой систематизации этих данных и их использование при отборе полиморфных локусов для анализа ассоциаций при генетико-эпидемиологических исследованиях многофакторных признаков (в том числе мультифакториальных заболеваний).
Цель исследования.
Провести систематический анализ данных, имеющихся в современной литературе о подходах к отбору полиморфных локусов при проведении ассоциативных исследований.
Материалы и методы.
В обзор включены современные данные зарубежных и отечественных статей, найденные в Pubmed, о подходах к отбору полиморфных локусов при проведении ассоциативных исследований многофакторных признаков (заболеваний).
Результаты исследования и их обсуждение.
Одним из основных требований при отборе полиморфного локуса для генетико-эпидемиологического исследования многофакторных признаков (заболеваний) является наличие его ассоциации с данным признаком (заболеванием), установленное в ранее проведенных исследованиях [3, 8, 33]. Основным ориентиром здесь являются данные полногеномных исследований (GWAS), результаты которых представлены в открытом доступе в каталоге полногеномных исследований (GWAS) National Human Genome Research Institute (http://www.genome.gov/gwastudies/). При этом в данном случае исследователь планирует проведение репликативного исследования в «своей» популяции [15], которая имеет своеобразие в генетических характеристиках («свою» историю формирования, миграционные потоки и т.д.), средовых условиях существования (климато-географические факторы и др.), кофакторах, которые играют значимую роль в развитии исследуемого фенотипа (заболевания) в данной выборке (например, артифициальные аборты являются фактором риска развития доброкачественных пролиферативных заболеваний матки у населения России, но не у населения Центральной Европы и Северной Америки и т.д.). Дополнительным аргументом для включения GWAS-значимого полиморфизма в исследование является подтверждение его значимой ассоциации в других GWAS исследованиях или независимых репликативных исследованиях. При отсутствии полногеномных исследований (например, GWAS исследования не проведены для гиперплазии эндометрия и ряда других заболеваний) или неоднозначности немногочисленных GWAS исследований отдельных многофакторных признаков (заболеваний) в исследование правомочно включать полиморфные локусы, которые, показали свою «значимость» в других, ранее проведенных ассоциативных исследованиях и в первую очередь «крупномасштабных» ассоциативных исследованиях (например, в работе He C. et al. [10], посвященной поиску ассоциаций генов-кандидатов с возрастом менархе для исследования были отобраны 18862 SNPs в 278 генах 9 различных функциональных групп. На выборке из 24341 женщин значимые ассоциации с возрастом менархе были установлены 42 SNPs 9 генов – FSHB, LHCGR, POMC, UGT2B4, GHRH, CD40LG, FGFR1, KISS1, NKX2-1. В работе Goddard K.A. et al. [14] при изучении ассоциации 775 SNP в 190 генах с развитием преэклампсии на выборке из 350 пар мать-ребенок с преэклампсией и 600 контрольных пар установлено 6 генов, ассоциированных с развитием ПЭ: IGF1, IL4R, IGF2R, GNB3, CSF1, THBS4).
При этом, следует отметить, что целый ряд многофакторных признаков и в т.ч. заболеваний характеризуются коморбидностью (достаточно часто встречается совместно) и синтропией (неслучайное сочетание двух и более болезней у индивидуума и его ближайших родственников, имеющее эволюционно-генетическую основу [5]. Синтропия определяется общими механизмами развития данных заболеваний и вовлеченностью в их формирование общих (синтропных) генов (функционально взаимодействующие ко-регулируемые гены, локализованные во всем пространстве генома человека, вовлеченные в общие для данной синтропии биохимические и физиологические пути) [6]. В соответствии с этим, при отборе полиморфных локусов для исследования следует учитывать наличие ассоциации «кандидатного» полиморфизма с фенотипами, имеющими с исследуемым признаком, общие биологические пути и вследствие этого имеющего для организма (как в норме, так и при развитии заболевания) более значимую – «ключевую» роль. Так, например, согласно данным литературы, среди генов-кандидатов менархе особое место занимают гены, ассоциированные также с антропометрическими характеристиками (рост, индекс массы тела и др.) (LIN28B, FTO, TNNI3K, MAP2K5, FANCL, STK33, GPRC5B,POMC/RBJ и др.), в связи с тем, что они вовлечены в общие биологические пути формирования менархе, роста, распределения жировой ткани, развития ожирения и др. [13, 22, 28]. Поэтому при изучении молекулярно-генетических факторов менархе для анализа ассоциаций предпочтительнее отбирать полиморфные локусы, которые так же показали в ранее проведенных исследованиях ассоциации с антропометрическими характеристиками.
При оценке функционального значения полиморфизма при его расположении в кодирующей части генома (экзонах) важно оценить является ли эта нуклеотидная замена несинонимической (т.е. приводит к замене аминокислоты в кодируемом полипептиде) и если это так, то необходимо понимать каков предикторный потенциал данной аминокислотной замены. Для выявления «миссенс-мутаций» (nsSNP) и оценке их предиктивного потенциала может использоваться программное обеспечение SIFT (Sorting Tolerant From Intolerant) [18] (http://sift.jcvi.org/) и PolyPhen-2 (Polymorphism Phenotyping v2) (prediction of functional effects of human nsSNPs) [11] (http://genetics.bwh.harvard.edu/pph2/index.shtml). В качестве примера использования этих он-лайн программ для оценки предикторного потенциала миссенс-замен приведем наши неопубликованные результаты изучения ассоциаций полиморфизма генов-кандидатов с развитием гиперплазии эндометрия. Нами установлено, rs4889 (1q32.1), сильно сцепленный с rs7538038 (r2=0.98), который ассоциирован с развитием гиперплазии эндометрия, обусловливает замену Pro81Arg в полипептиде KISS1. Согласно базы данных PolyPhen-2 (prediction of functional effects of human nsSNPs) аминокислотная замену Pro81Arg в полипептиде KISS1 имеет предикторный класс «POSSIBLY DAMAGING» с score=0.524 (чувствительность 0.88, специфичность – 0.90). Так же, нами показано, что rs4633 (ассоциирован с гиперплазией эндометрия и приводит к синонимичной замене аминокислоты в белковом продукте гена COMT, 22q11.21) сильно сцеплен (r2=0.99) с rs4680, который определяет замену Val18Met в полипептиде COMT. Согласно данных базы SIFT (Sorting Tolerant From Intolerant) аминокислотная замена Val18Met в полипептиде COMT имеет SIFT Score=0.02, что характеризует ее предикторное значение как «DELETERIOUS» (SIFT Score ≤0.05). Следует отметить, что в ранее проведенной работе Chen J. et al. [17] показано, что замена Val/Met в полипептиде COMT обусловливает изменение стабильности и активности COMT: Met вариант приводит к 40% снижению уровня белка и уменьшению его ферментативной активности по сравнению с вариантом Val. Другой пример демонстрирующий важность оценки предикторного потенциала полиморфизма, несмотря на то, что он является несинонимическим – нами было установлено (неопубликованные данные) что с развитием генитального эндометриоза ассоциирован полиморфизм rs713586 (2p23.3) с которым сильно сцеплен (r2=0.93) rs11676272, обусловливающий замену Ser107Pro в полипептиде ADCY3. Эта аминокислотная замена имеет предикторное значение «TOLERATED» по базе данных SIFT с SIFT Score=0.42, что не превышает порогового значения SIFT Score ≤0.05. Также, по базе данных PolyPhen-2 данная аминокислотная замена имеет предикторный класс «BENIGN» с score=0 (чувствительность 1.00, специфичность – 0). Таким образом, хотя полиморфный локус rs11676272 и обусловливает миссенс замену в полипептиде ADCY3, однако она не влияет на функциональное значение этого полипептида.
Важное значение при характеристике полиморфного локуса имеет его регуляторный потенциал и eQTL значение (связь с экспрессией генов). Регуляторный потенциал полиморфных локусов, а также их связь с экспрессией генов, все чаще исследователями оцениваются не только в post-hoc анализе результатов полногеномных исследований (пример такого post-hoc анализа функционального значения некодирующих GWAS-значимых полиморфных локусов представлен в виде блок-схемы на рис. 1) или ассоциативных исследований отдельных групп генов-кандидатов, а при планировании генетико-эпидемиологических исследований с целью отбора функционально значимых полиморфных локусов. В современной литературе представлено достаточно много мировых баз данных по функциональной геномике и биоинформатических методов анализа этих данных, позволяющих insilico оценить регуляторный потенциал и eQTL значение SNPs, планируемых для исследования. В таблицах 1 и 2 представлена в качестве примера информация о публично доступных информационных ресурсах для оценки регуляторного потенциала (табл. 1) и влияния на экспрессию генов (табл. 2) изучаемых (или планируемых для исследования) полиморфных локусов, взятая из работы Tak Y.G. и Farnham P.J. [26] «Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non‑coding regions of the human genome», опубликованной в журнале «Epigenetics&chromatin». Следует отметить, что в современные онлайн ресурсы по функциональной геномике достаточно хорошо интегрированы данные, полученные в ходе реализации крупных международных проектов в области эпигенетики – ENCODE (The Encyclopedia of DNA Elements) (https://www.encodeproject.org/) и консорциума NIH Roadmap Epigenomics Mapping Consortium (http://www.roadmapepigenomics.org/). С помощью этих данных оценивается расположение полиморфизма в регионе «открытого хроматина», регионах модифицированных гистонов, сайтах связывания с различными регуляторными белками (CTCF и др.), регионе мотивов ДНК, являющихся сайтами связывания с транскрипционными факторами (TFBs) и т.д. (рис. 2) [20], и результатом этого является приоритизация SNPs с позиций их регуляторных эффектов (рис. 3) [26].
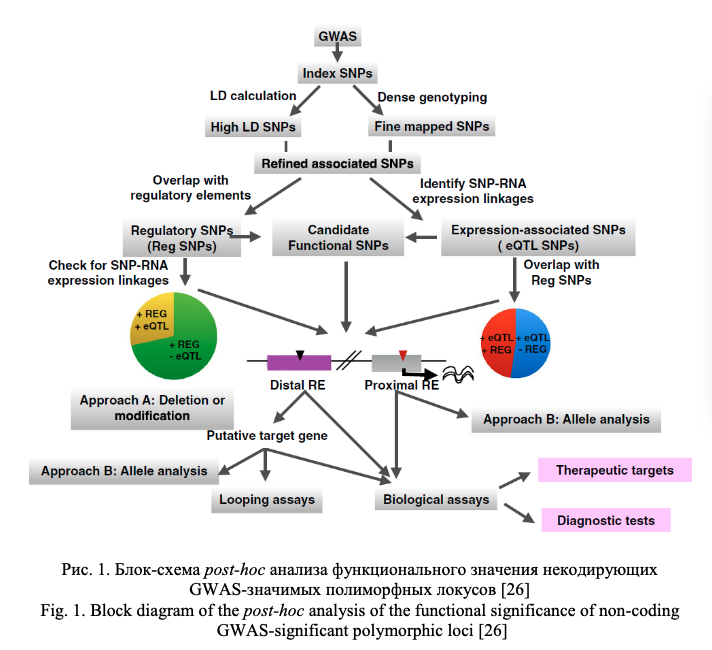
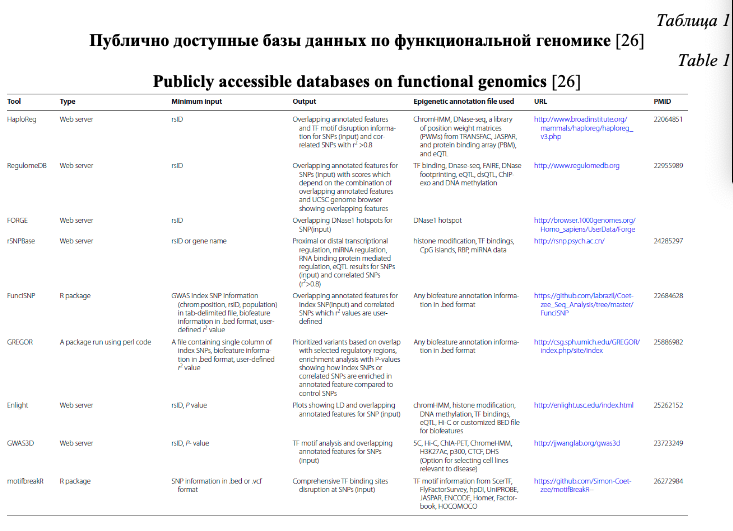
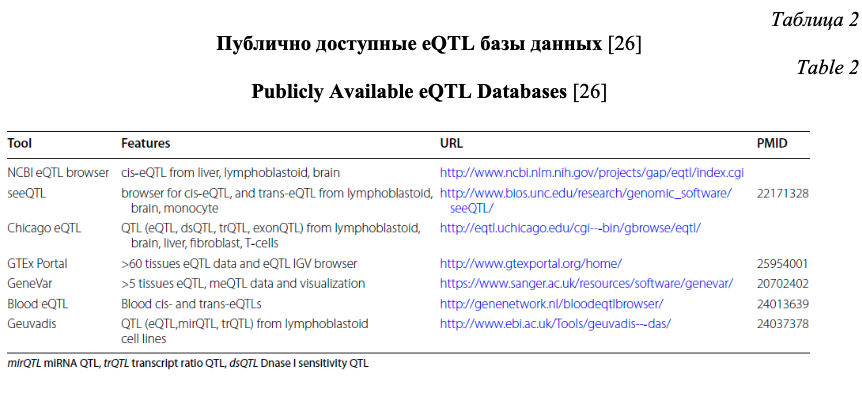
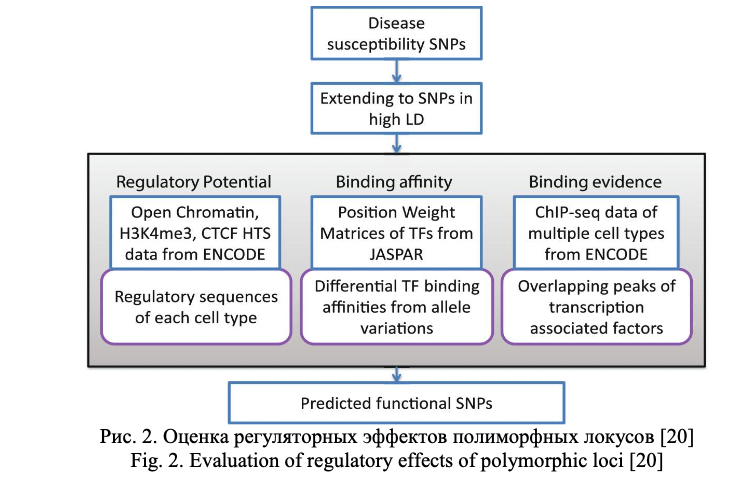
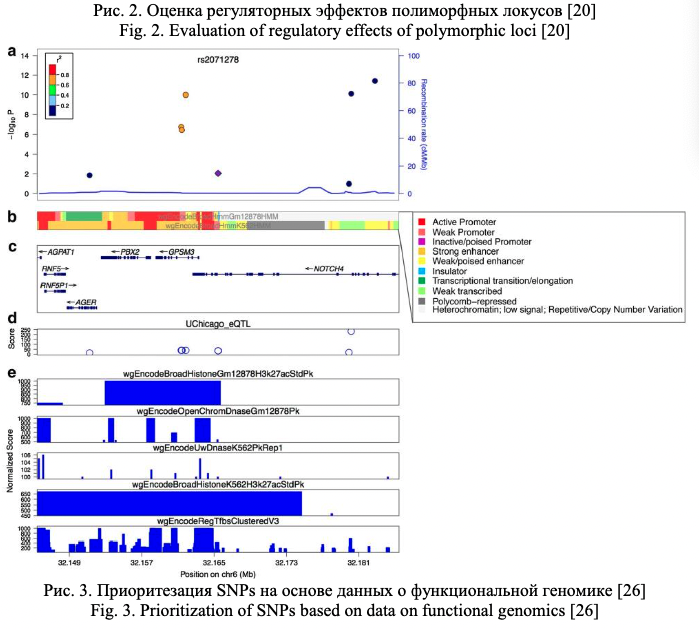
Онлайн ресурсы (RegulomeDB, SNP Function Prediction, rSNPBase и др.) широко используются при планировании генетико-эпидемиологических исследований в работах различных научных коллективов и в том числе отечественными учеными [1, 7, 9, 16, 19, 29].
Нашим научным коллективом, при планировании генетико-эпидемиологического исследования возраста менархе, для оценки регуляторного потенциала полиморфных локусов, отбираемых для ассоциативного анализа использовалось следующее онлайн программное обеспечение:
- HaploReg (v4.1) [30] (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php) (Core 25-state model using 12 imputed marks) (рассматривается расположение SNPs в области гистонов, маркирующих промоторы и энхансеры, в регионе гиперчувствительности к ДНКазе, эволюционно консервативных регионах, регионе регуляторных мотивов и регионе связывания с регуляторными белками);
- rSNPs MAPPER (Multi-genome Analysis of Positions and Patterns of Elements of Regulation) [23] (http://genome.ufl.edu/mapper/mapper-main) (оценивается локализация SNPs в регионе сайтов связывания с транскрипционными факторами на основе баз данных TRANSFAC, JASPAR, MAPPER);
- RegulomeDB (Version 1.1) [12] (http://regulome.stanford.edu/) (оценивается regulatory scores SNPs). В онлайн ресурсе RegulomeDB для SNP представлены регуляторные коэффициенты – 1a (eQTL + TF binding + matched TF motif + matched DNase Footprint + DNase peak), 1b (eQTL + TF binding + any motif + DNase Footprint + DNase peak), 1c (eQTL + TF binding + matched TF motif + DNase peak), 1d (eQTL + TF binding + any motif + DNase peak), 1e (eQTL + TF binding + matched TF motif), 1f (eQTL + TF binding / DNase peak), 2a (TF binding + matched TF motif + matched DNase Footprint + DNase peak), 2b (TF binding + any motif + DNase Footprint + DNase peak), 2c (TF binding + matched TF motif + DNase peak), 3a (TF binding + any motif + DNase peak), 3b (TF binding + matched TF motif), 4 (TF binding + DNase peak), 5 (TF binding or DNase peak), 6 (other). Следует отметить, что чем меньше коэффициент, тем выше регуляторный потенциал SNP и влияние на экспрессию генов оказывают только SNPs, имеющие коэффициент от 1а до 1f;
- rSNPBase [24] (http://rsnp.psych.ac.cn/index.do) (оценивается роль SNPs в проксимальной и дистальной транскрипционной регуляции, регуляции, опосредованной РНК-связываемыми
белками); - SNPinfo Web Server – SNP Function Prediction (FuncPred) [32] (https://snpinfo.niehs.nih.gov/snpinfo/snpfunc.html) (оценивается Regulatory Potential Score SNPs, а также связь полиморфизма с сайтами, связывающими факторы транскрипции (TFBs), микроРНК, консервативными последовательностями и др.).
С помощью современных баз данных по функциональной геномике можно оценить конкретную связь полиморфного локуса (референсного и альтернативного аллелей) с изменением регуляторного мотива ДНК (афинность мотива к транскрипционным факторам). Одним из таких подходов является определение разницы между LOD scores альтернативного (alt) и референсного (ref) аллелей [22, 31]: LOD (alt) − LOD (ref). Отрицательное значение этого показателя свидетельствует о повышении афинности данного мотива референсным аллелем, наоборот, положительное значение демонстрирует связь альтернативного аллеля с повышением афинности анализируемого мотива ДНК.
При рассмотрении функционального значения полиморфного локуса, включаемого в исследование, наряду с его регуляторными эффектами следует оценивать и его связь с экспрессией генов (eQTL значение). В таблице 2 представлена информация о публично доступных eQTL базах данных, позволяющих оценить связь полиморфизма с экспрессией генов [26]. В качестве примера остановимся на характеристиках некоторых из них. С помощью данных онлайн программы Blood eQTL browser [25] (http://genenetwork.nl/bloodeqtlbrowser/), основанных на eQTL анализе нетрансформированных образцов периферической крови 5311 индивидуумов, могут быть оценены ассоциации исследуемых SNPs с уровнем экспрессии генов в периферической крови (cis- и trans-eQTL). При локализации SNP от гена на расстоянии до 250 kb его эффекты оценивались как cis-eQTL, а при расположении полиморфизма от гена на дистанции более чем 5Mb – как trans-eQTL. Для оценки значимости полученных данных используется показатель False Discovery Rate (FDR). Статистически значимыми считаются данные с уровнем значимости p<5*10-5 , pFDR≤0.05.
С использованием данных проекта Genotype-Tissue Expression (GTEx) [27], основанных на результатах анализа экспрессии генов (341316 eGenes – eQTL для 27159 уникальных генов) в 10294 образцах из 48 органов и тканей (не менее 70 образцов из каждого органа или ткани) 620 индивидуумов и представленных в онлайн программе GTExportal на 10.12.2017 (Release V7 updated on 09/05/2017) (dbGaP Accession phs000424.v7.p2) (http://www.gtexportal.org/), может быть изучен экспрессионный потенциал (cis-eQTL) исследуемых SNP в различных органах и тканях и в том числе патогенетически значимых для развития данного многофакторного признака (заболевания). Для идентификации значимых eQTL применяется показатель False Discovery Rate (FDR) (статистически значимыми считаются результаты с p<8*10-5, pFDR≤0.05). С помощью данного онлайн сервиса можно оценить влияние аллельных вариантов рассматриваемого полиморфизма на уровень экспрессии определенного гена – на основе оценки коэффициента линейной регрессии (β), характеризующего изменение нормализованного показателя генной экспрессии на один полиморфный (альтернативный) генетический вариант.
Следует отметить, что при выборе полиморфных локусов для исследования особое внимание следует уделять рассмотрению вопроса о функциональной роли полиморфизма данного гена-кандидата в органе или ткани, вовлеченных в формирование исследуемого многофакторного признака (заболевания), т.е. рассматривать регуляторный потенциал SNPs с учетом дифференциальной экспрессии генов-кандидатов в органах (тканях) - мишенях. Данный подход к отбору полиморфных локусов для ассоциативного анализа в РФ активно развивается научным коллективом из Томского НИИ медицинской генетики под руководством член-корр. РАН В.А. Степанова. Используя данный подход, ученые из этого научного коллектива отобрали для генетико-эпидемиологического исследования преэклампсии 46 регуляторных полиморфных вариантов 21 дифференциально экспрессируемых в плаценте генов-кандидатов [1, 7]. Как указывают авторы, примененный ими новый подход поиска генетических маркеров мультифакториальных заболеваний, основанный на комбинации геномных, транскриптомных и биоинформатических подходов (заключается в выборе дифференциально экспрессируемых генов на основании полногеномного анализа транскриптома плацентарной ткани по результатам собственного исследования и ранее опубликованных данных, биоинформатического поиска полиморфных маркеров в регуляторных участках этих дифференциально экспрессируемых генов, анализе ассоциаций с преэклампсией методом случай-контроль) способен обнаружить новые потенциальные генетические маркеры в генах, предположительно вовлеченных в патогенез заболевания, которые, вероятно, входят в состав “упущенной наследуемости” при мультифакториальных заболеваниях и не могут быть выявлены при геномном анализе [1].
Следует отметить, что при оценке функционального значения SNP важно также учитывать не только его самостоятельные эффекты (эпигенетические эффекты данного полиморфизма и его связь с экспрессией генов) но и функциональное значение полиморфных локусов, находящихся с ним в неравновесии по сцеплению (при выделении определенного уровня силы сцепления r2≥0.2-0.8) т.к. именно эти сильно сцепленные с рассматриваемым SNP полиморфные локусы, как правило, будут демонстрировать наиболее значимые регуляторные эффекты в сравнении с функциональными эффектами этого одного SNP. Так, например, с отобранными нами для генетико-эпидемиологического исследования возраста менархе у населения России 52 полиморфными локусами оказались сильно сцеплены (r2≥0.8) около 1000 SNPs (n=980), при этом их регуляторные эффекты, влияние на экспрессию генов, связь с несинонимическими заменами были существенно выше нежели 52 SNPs, изначально включенных в ассоциативное исследование (наши неопубликованные данные).
В настоящее время одной из наиболее продуктивных стратегий создания панели генетических маркеров для анализа ассоциаций с мультифакториальными заболеваниями является выбор tagger SNP (tagSNP) с целью расширения «генетического охвата» [2]. Как указывается в работе Трифоновой Е.А. и др. [2], подход с использованием tagSNPs имеет преимущества в связи с известными на сегодняшний день знаниями о блочной структуре LD организации генома человека и существенно редуцирует силы и средства, необходимые для генотипирования. В работе Хаджиевой М.Б. и др. [4] отмечается, что стратегия применения tagSNPs основана на блочной структуре генома, каждый такой блок включает в себя соседние или близко расположенные аллели, наследуемые совместно чаще, чем это можно ожидать при случайном распределении; данное явление известно как неравновесное сцепление (linkage disequilibrium, LD). SNP с частотой минорного аллеля (MAF) не менее 5% маркирует соответствующий гаплотипический блок при условии, что он находится в пороговой корреляции хотя бы с одним SNP из данного блока (D' ≥ 0.8). Исследование tagSNPs из каждой группы сцепления обеспечивает покрытие всего гена.
Подбор tagSNPs может быть выполнен в программе HaploView (version 4.2) (https://www.broadinstitute.org/haploview/haploview), которая определяет гаплотипическую структуру гена для конкретной популяции, основываясь на данных проекта HapMap (haplotype map) (http://hapmap.ncbi.nlm.nih.gov/). Наряду с этим tagSNPs могут быть оценены с помощью программного обеспечения SNPinfo Web Server National Institute of Environmental Health Sciences (NIEHS) – LD TAG SNP Selection (TagSNP) (https://snpinfo.niehs.nih.gov/snpinfo/snptag.html) с использованием данных International HapMap (phase III) и базы dbSNP для различных популяций на основе оценки неравновесия по сцеплению (LD) между парами SNPs (при максимальной дистанции между SNP для определения LD 250kb) с заданным порогом r2 (например, r2≥0.8). В настоящее время данная стратегия выбора полиморфных маркеров для анализа ассоциаций полиморфизма с развитием мультифакториальных заболеваний активно используется различными коллективами ученых и в том числе отечественными генетиками [2, 4].
В исследовании Трифоновой Е.А. и др. [2] в качестве маркеров для изучения паттернов LD были выбраны 12 SNPs ген метилентетрагидрофолатредуктазы (MTHFR), относительно равномерно охватывающие все участки гена (экзоны, интроны, 3'-нетранслируемые участки) при частоте минорного аллеля в большинстве локусов не менее 5% (по данным базы NСBI). Для выделения tagSNPs авторы использовали два метода – «STAMPA» и «Tagger». Согласно полученным авторами результатам, при заданном пороге прогностической ценности в различных популяциях наблюдаются неодинаковые наборы tagSNPs, что, по мнению авторов, вероятно, обусловлено варьированием структуры LD и гаплотипического разнообразия гена MTHFR в выборках. Вопрос о возможности «переноса» tagSNPs с одной популяции на другую довольно важен, так как анализ ассоциаций с использованием tagSNPs, идентифицированных на основании данных проекта HapMap, весьма актуален. Авторы отмечают, что качество выбранных tagSNPs зависит от исходного массива, в котором они охарактеризованы. Если изначально плотность маркеров слишком редкая, то выбранный tagSNP «захватит» меньше информации, чем требуется для анализа. Необходимая плотность маркеров в исходном массиве данных варьирует в различных участках генома в зависимости от ряда показателей, таких, например, как уровень рекомбинации, структура LD, частоты SNP, характер мутации и демографическая история населения [2]. Следует отметить, что на выбор tagSNP наряду с количеством анализируемых SNP (плотность маркеров) значимое влияние оказывают используемый при этом алгоритм выделения гаплоблоков («Confidence intervals», «Solid spine of LD» и др.) и выбранный порог оценки неравновесия по сцеплению
(D' ≥ 0.75, D' ≥ 0.80 и др.)
В работе Хаджиевой М.Б. и др. [4], посвященной изучению полиморфизма генов, контролирующих процессы эластогенеза, и риска развития пролапса тазовых органов у женщин, в связи с отсутствием для генов FBLN3 и LOXL1 известных функциональных полиморных вариантов, был использован подход, заключающийся в подборе таргетных (tagger) SNPs. Авторы с помощью программы HaploView (version 4.2) используя данные для популяций CEU + TSI (CEU – американцы европейского происхождения, TSI – жители Тосканы) из проекта “HapMap” отобрали для исследования 3 tagSNPs гена LOXL1 (rs2165241, rs2304719, rs893821) и 4 SNPs гена FBLN3 (rs3791679, rs1367228, rs3791660 и rs2033316), из которых все, кроме rs1367228, являются таргетными.
Таким образом, согласно современным представлениям при отборе полиморфных локусов генов-кандидатов для изучения ассоциаций с мультифакториальным признаком (заболеванием) следует учитывать следующие критерии: 1) наличие ассоциации с исследуемым признаком по результатам ранее проведенных полногеномных (GWAS) и/или ассоциативных (в том числе репликативных) исследований; 2) наличие ассоциации с фенотипами, имеющими с исследуемым признаком, общие биологические пути; 3) регуляторный потенциал (regSNP);
4) влияние на экспрессию генов (eSNP);
5) связь с несинонимическими заменами (nsSNP); 6) tagger SNP (tagSNP) 7) частота полиморфизма не менее 5% 8) функциональные эффекты (regSNP, eSNP, nsSNP) SNPs, находящихся в неравновесии по сцеплению (r2≥0.8) с отобранными для ассоциативного анализа полиморфизмами.
Похожие статьи
Инна Николаевна Сорокина
Наталья Александровна Рудых
Ирина Николаевна Безменова
Ирина Сергеевна Полякова
и другие.
Ирина Васильевна Пономаренко
Список литературы