Эта статья опубликована под лицензией Creative Commons и не автором статьи. Поэтому если вы найдете какие-либо неточности, вы можете исправить их, обновив статью.

Diversity and community structure of bacterioplankton in surface waters off the northern tip of the Antarctic Peninsula



Shunan Cao
Jianfeng He
Fang Zhang
Ling Lin
Yuan Gao
Qiming Zhou
Опубликована Март 21, 2019
Последнее обновление статьи Авг. 6, 2023
Эта статья опубликована под лицензией
")



Abstract
Global climate change is significantly affecting marine life off the northern tip of the Antarctic Peninsula, but little is known about microbial ecology in this area. The main goal of this study was to investigate the bacterioplankton community structure in surface waters using pyrosequencing and to determine factors influencing this community. Pelagibacterales and Rhodobacterales (Alphaproteobacteria), Oceanospirillales and Alteromonadales (Gammaproteobacteria), and Flavobacteriales (Bacteroidetes) were the core taxa in our samples, and the five most relatively abundant genera were Pelagibacter, Polaribacter, Octadecabacter, group HTCC2207 and Sulfitobacter. Although nutrients and chlorophyll a (chl a) contributed more to bacterioplankton community structure than water masses or depth, only 30.39% of the variance could be explained by the investigated environmental factors, as revealed by RDA and pRDA. No significant difference with respect to nutrients and chl a was observed among water masses or depth, as indicated by ANOVA. Furthermore, significant correlations among the dominant bacterial genera were more common than correlations between dominant genera and environmental factors, as revealed by Spearman analysis. We conclude that nutrients and chl a become homogeneous and that interpopulation interactions may have a central role in influencing the bacterial community structure in surface waters off the northern tip of the Antarctic Peninsula during the summer.
Ключевые слова
Dominant genera, pyrosequencing, interpopulation interactions, Bacteria, environmental factors
Introduction
The Southern Ocean plays a vital role in global ecosystem functions, such as influencing primary production (Ducklow 1999), sustaining the marine food web and contributing to biogeochemical cycles (Azam 1998; Azam & Malfatti 2007; Luria et al. 2014). The Southern Ocean occupies approximately 10% of the total global ocean surface area and accounts for about 30% of the oceanic CO2 uptake (Wilkins, Yau et al. 2013). Polar environments such as the Southern Ocean are especially sensitive to global warming (Loeb et al. 1997; Moline et al. 2004). The glaciers of the southern Antarctic Peninsula are diminishing (Wouters et al. 2015), and the ice shelves of the northern region are retreating along the coast (Cook & Vaughan 2010); both these changes could have important impacts on the Antarctic marine ecosystem. The polar marine ecosystem rapidly responds to climate change (Moline et al. 2009; Doney et al. 2012) and, as a consequence, it is experiencing shifts in the food web, from the base to higher trophic levels (Moline et al. 2004; Kirchman et al. 2009; Montes-Hugo et al. 2009; Schofield et al. 2010; Signori et al. 2018).
In these waters, bacterioplankton has an approximate abundance of 108 cells L−1; however, they live in low water temperature and present seasonal variations in productivity (Delille 2004; Manganelli et al. 2009). Alphaproteobacteria, Gammaproteobacteria and CFB are the most dominant bacterioplankton groups, followed by Betaproteobacteria, Deltaproteobacteria, Verrucomicrobia, Planctomycetes, Actinobacteria and Epsilonproteobacteria (Wilkins, Yau et al. 2013).
The bacterioplankton community structure may be influenced by various factors, not only environmental, including interaction with other microbial groups. Seasonal darkness and sea-ice coverage prevent photoautotrophy and cause the bacterioplankton community to change seasonally. In February (summer), chemoheterotrophic and photoheterotrophic bacteria are dominant, whereas in August (winter) chemolithoautotrophic bacteria are more abundant (Grzymski et al. 2012). Furthermore, bacterioplankton assemblages can differ with water depth, or be unique to different water masses (Luria et al. 2014; Signori et al. 2014), as depth and water mass may represent unique physical/chemical conditions. For example, across the Bransfield Strait (north-western Antarctic Peninsula), Bacteroidetes and Proteobacteria (mainly Alpha- and Gammaproteobacteria) dominate the mixed layer above 100 m, while Thaumarchaeota, Euryarchaeota and Proteobacteria (Gamma-, Delta-, Beta- and Alphaproteobacteria) are the main taxonomic groups below 100 m (Signori et al. 2014). In the WAP, winter bacterial assemblages most closely resembled summer assemblages from the sub-euphotic zone. The relative abundance of some Proteobacteria clusters was significantly greater at 100 m than at 10 m, reflecting the well-established seasonal patterns of water column turn-over and stratification that results in an isolated layer of “winter water” below the euphotic zone (Luria et al. 2014). In addition to environmental factors, the presence of Archaea and Eukarya can influence the bacterioplankton community structure, as suggested by the reported association between diatoms and Rhodobacteraceae, Cryomorphaceae and SAR11 (Luria et al. 2014).
Marine ecosystems of the western and northern Antarctic Peninsula, a region that is experiencing rapid regional warming, have been receiving considerable research attention recently (Church et al. 2003; Anisimov et al. 2007; Ducklow et al. 2012; Grzymski et al. 2012; Luria et al. 2014; Signori et al. 2014; Luria et al. 2016; Bowman et al. 2017; Signori et al. 2018). However, less attention has been given to the waters off the northern tip of the Antarctic Peninsula, which includes the Powell Basin, the eastern South Shetland Islands and the South Orkney tableland, lying between the Scotia Sea and the Weddell Sea. This area not only represents the confluence site of waters coming from the Weddell and Scotia seas but is also a region of water mass transformation under the influence of climate change (Gordon 1967; Meredith et al. 2014).
Investigating the bacterioplankton diversity and community structure in the waters off the northern tip of the Antarctic Peninsula will help us to better understand the marine ecosystem. More studies addressing the bacterioplankton taxonomic composition at the genus and species level, rather than at higher taxonomic levels, such as phyla and classes, are needed to provide further details on taxonomic shifts and associations within different marine ecosystems (Agogué et al. 2011).
To better understand the environmental and biological factors influencing bacterioplankton diversity and community structure in this underexplored region of the Southern Ocean, we selected sampling stations along transects between the Scotia Sea and Weddell Sea. Bacterioplankton communities were assessed by amplicon pyrosequencing of the 16S rRNA genes, which were further correlated with nutrient, chl a, in situ temperature and salinity measurements. Our work constitutes a baseline for future research in this region.
Materials and Methods
Study area and sampling strategy
Samples were collected in the waters by the northern tip of the Antarctic Peninsula, including the northern area of the Bransfield Strait, the Powell Basin and the South Orkney tableland area (Fig. 1). The SSW from the Scotia sea and the Weddell Sea (Gordon & Huber 1995) and WW from the Scotia Sea represent upper waters. This research was conducted during the 28th Chinese National Antarctic Research Expedition, from December 2011 to January 2012.
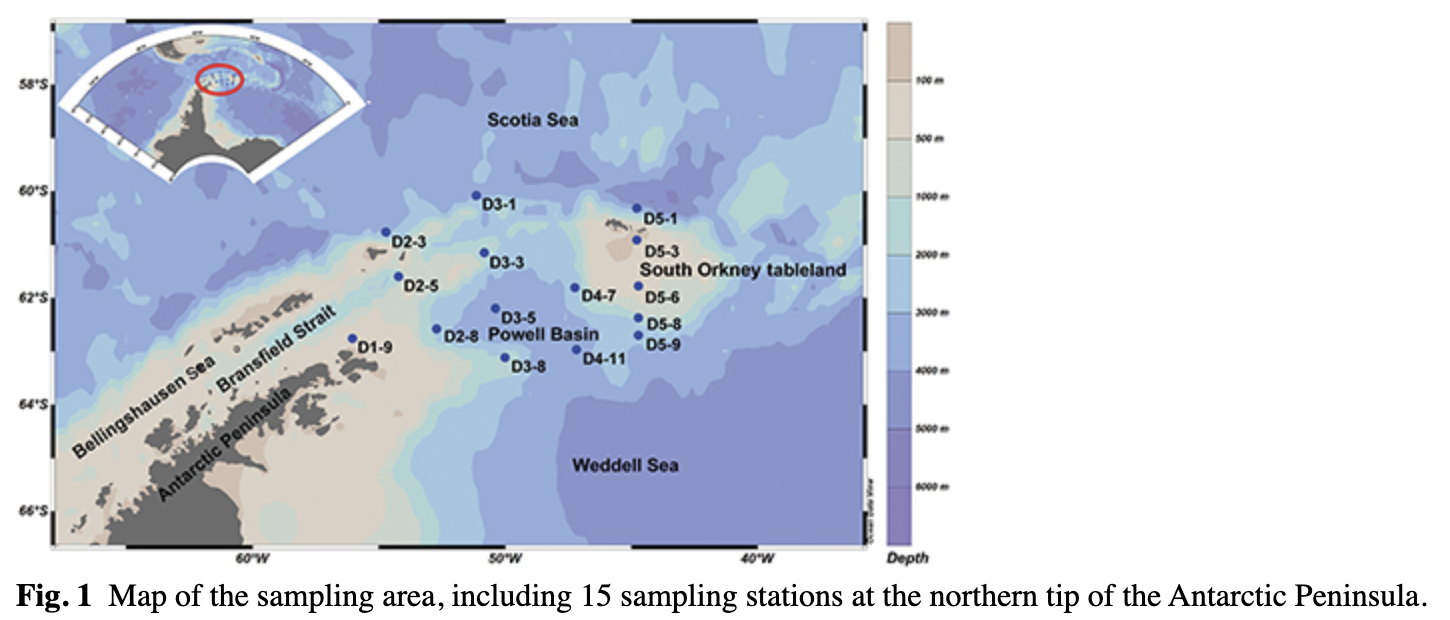
A total of 18 samples were collected along the transects between the Scotia and Weddell seas (Fig. 1). Thirteen samples were taken at 25 m depth (the layer of maximum oxygen), three samples were taken at 2 m and two samples were taken at 50 m (Table 1). An SBE 911 plus CTD instrument combined with an SBE 32 Carousel water sampler (both by Sea-Bird Electronics) equipped with 24 Niskin bottles was used to collect seawater and measure physical parameters (temperature and salinity). Sampling depth was confirmed by the CTD profiles. Approximately 2 L of seawater was filtered with the assistance of a vacuum pump through polycarbonate membranes (47 mm diameter, 0.2-μm pore size) following pre-filtration through 3 μm pore size polycarbonate membranes (all membranes by Whatman). The membranes were immediately frozen at −80°C and shipped to the laboratory for microbial diversity analysis.
Table 1 Environmental parameters and locations of each sampling station. | ||||||||||||
Station | Depth | Date | Longitude (W) | Latitude (S) | Temperature (°C) | Salinity (psu) | Nitrite (μmol/L) | Nitrate (μmol/L) | Ammonium (μmol/L) | Phosphate (μmol/L) | Silicate (μmol/L) | Chl a (μg/L) |
D1-9 | 2 ma | 2012-01-17 | 55°59.515′ | 62°44.912′ | −0.13 | 34.43 | 0.12 | 53.03 | 1.06 | 2.27 | 71.61 | 0.51 |
D1-9 | 50 ma | 2012-01-17 | 55°59.515′ | 62°44.912′ | −0.18 | 34.43 | 0.25 | 49.54 | 1.12 | 1.96 | 69.77 | 1.03 |
D2-3 | 25 mb | 2012-01-23 | 54°40.857′ | 60°48.085′ | 0.95 | 34.30 | 0.22 | 29.82 | 1.33 | 2.03 | 70.44 | 0.51 |
D2-5 | 2 mb | 2012-01-23 | 54°08.834′ | 61°35.359′ | 0.63 | 34.32 | 0.22 | 46.94 | 1.25 | 1.92 | 70.44 | 0.74 |
D2-5 | 25 mb | 2012-01-23 | 54°08.834′ | 61°35.359′ | 0.62 | 34.32 | 0.27 | 47.53 | 1.22 | 1.96 | 72.68 | 0.85 |
D2-8 | 25 mc | 2012-01-20 | 52°44.032′ | 62°34.216′ | 0.33 | 34.11 | 0.10 | 47.14 | 1.91 | 1.55 | 68.51 | 0.23 |
D3-1 | 25 mb | 2012-01-24 | 51°9.137′ | 60°06.009′ | 1.16 | 34.27 | 0.18 | 32.39 | 0.97 | 2.20 | 67.42 | 0.71 |
D3-3 | 25 ma | 2012-01-24 | 50°45.609′ | 61°09.446′ | −0.04 | 34.50 | 0.11 | 38.16 | 0.93 | 2.34 | 87.10 | 0.21 |
D3-5 | 25 mb | 2012-01-25 | 50°21.610′ | 62°13.943′ | 0.49 | 34.06 | 0.13 | 27.45 | 1.30 | 1.783 | 75.41 | 0.20 |
D3-8 | 25 md | 2012-01-26 | 49°57.468′ | 63°8.767′ | −1.30 | 33.70 | 0.20 | 28.66 | 1.76 | 1.87 | 68.88 | 0.08 |
D4-7 | 25 mb | 2012-01-27 | 47°10.694′ | 61°47.881′ | 0.48 | 34.27 | 0.34 | 28.84 | 0.50 | 1.80 | 66.54 | 0.79 |
D4-11 | 25 md | 2012-01-26 | 47°9.644′ | 62°58.519′ | −0.52 | 33.58 | 0.16 | 25.04 | 1.97 | 1.55 | 77.94 | 0.25 |
D5-1 | 25 mc | 2012-01-28 | 44°41.800′ | 60°19.012′ | 0.37 | 34.00 | 0.21 | 29.17 | 1.13 | 1.74 | 66.25 | 0.36 |
D5-3 | 2 ma | 2011-12-30 | 44°42.320′ | 60°55.062′ | −0.02 | 34.14 | 0.24 | 23.20 | 1.53 | 1.48 | 68.80 | 2.14 |
D5-3 | 25 ma | 2011-12-30 | 44°42.320′ | 60°55.062′ | −0.46 | 34.15 | 0.25 | 27.26 | 1.25 | 1.78 | 71.31 | 2.11 |
D5-6 | 25 mc | 2012-01-29 | 44°40.989′ | 61°47.396′ | 0.39 | 34.27 | 0.18 | 40.95 | 0.78 | 1.83 | 74.33 | 0.43 |
D5-8 | 25 ma | 2012-01-29 | 44°41.432′ | 62°23.262′ | −0.18 | 33.95 | 0.24 | 29.65 | 1.12 | 1.90 | 73.65 | 0.22 |
D5-9 | 50 mc | 2012-01-29 | 44°41.354′ | 62°42.042′ | −1.02 | 34.19 | 0.34 | 33.06 | 0.88 | 2.06 | 75.02 | 0.22 |
aWWs. bSSWs. cMWM. dSSWw. | ||||||||||||
Inorganic nutrients and chl a concentrations
Subsamples (100 mL) for determination of inorganic nutrient (nitrate, nitrite, silicate and phosphate) concentrations were filtered through a 0.45-μm pore size, 47-mm diameter Whatman polycarbonate membrane. Nutrients were determined using an autoanalyser (Skalar San++), using methods explained by Grasshoff et al. (2007) and He et al. (2012).
Subsamples (500 mL) for chl a determination were passed through 47-mm diameter Whatman GF/F filters under low vacuum pressure and analysed onboard the research vessel using a fluorometer (Turner Designs 10), in accordance with the methods of Parsons et al. (1984) and He et al. (2012).
Total DNA extraction, amplification and sequencing
Total DNA was extracted using a modified cetyltrimethylammonium bromide method (Zhang et al. 2015) and examined by agarose gel electrophoresis. The V1–V3 hypervariable region of the bacterial 16S rRNA gene was amplified using the universal primer pair F8 (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-AGAGTTTGATCCTGGCTCAG-3′) (Turner et al. 1999) and R533 (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-NNNNNNNN-TTACCGCGGCTGCTGGCAC-3′) (Sun et al. 2014). The adaptor sequences are italicized and the sample-specific barcode sequence is denoted by NNNNNNNN. PCR was performed using 5–10 ng genomic DNA in a final volume of 50 μL. The PCR procedure was as follows: initial denaturation at 95°C for 2 min; 25 cycles at 95°C for 30 s, 56.4°C for 1 min and 72°C for 30 s; and a final extension at 72°C for 5 min. The PCR products were purified using AxyPrepTM DNA Purification Kit (Axygen®) and quantified using a Qubit® 2.0 Fluorometer (Life Technologies). Pyrosequencing was performed on an FLX Titanium Genome Sequencer (454/Roche Life Sciences).
Partial 16S rRNA sequence data and statistical analyses
Mothur 3.1.2 (Schloss et al. 2009) was used to process partial raw 16S rRNA gene sequences. Reads were filtered using the following parameters: length (400 ≤ length ≤ 650 bp), quality score (≥ 25), number of ambiguous bases (= 0) and length of homopolymer runs (< 8). Sequences were clustered at 97% similarity using the SILVA database (version silva.seed_v119; Pruesse et al. 2007) as reference sequences. Taxonomy was assigned to OTUs using the Greengenes 13_5 database (McDonald et al. 2012). Chimeras, as well as sequences assigned to mitochondria and chloroplasts, were removed from the data set. The chloroplast sequences were annotated by both SILVA (silva.seed_v119; Pruesse et al. 2007) and Greengenes 13_5 (McDonald et al. 2012). Multiple rarefactions with 5738 reads per sample were performed for α-diversity analysis based on random sampling-based normalization.
R software (version 3.1.2) was used to determine α-diversity indices (ACE, Chao, Shannon, Inverse Simpson and Good’s coverage) based on the normalized sequence data (5738 reads per sample). Co-occurrence between environmental factors and bacterial communities was analysed by RDA after calculation by DCA (the largest axis length was 1.0089, which was < 3) (Lepš & Šmilauer 2003) (Supplementary Table S1). DCA, RDA and pRDA were performed at the OTU level using the vegan package in R (Oksanen et al. 2018). Pairwise comparisons of environmental factors and bacterial communities with a colour gradient denoting Spearman’s correlation coefficients and cluster analysis were also conducted using R.
Spearman correlation analysis (SPSS 20.0, IBM ETC) was used to investigate the significance of correlations between the bacterial genera with relative abundances >0.1% of the total data set and various environmental factors, including temperature, salinity, water masses, inorganic nutrients and chl a.
Differences in nutrients, chl a concentrations and in α-diversity indices were investigated between groups of samples, such as different depths and water masses, by ANOVA at the genus level using SPSS 20.0.
The sampling map was generated by the software Ocean Data View (Schlitzer 2018) and modified with Photoshop CS5 (Adobe).
Results
Environmental parameters
Four water masses were distinguished using in situ measurements data of temperature and salinity. These were the SSWs (T > 0.5°C, S > 34 psu), SSWw (−0.5°C > T >–1.5°C, S < 34 psu; Gordon & Huber 1995) and the WWs (−0.5°C < T < 0°C; Shi et al. 2016; Fig. 2). Other samples were assigned as the fourth water mass, referred to as the MWM (Table 1).

For each sampling site off the northern tip of the Antarctic Peninsula, the concentrations of nutrients (including nitrite, nitrate, ammonium, phosphate and silicate) and chl a were measured (Table 1). Mean concentrations and minimum–maximum ranges were obtained for nitrite (0.21 μmol L−1, 0.10–0.34 μmol L−1); nitrate (35.44 μmol L−1, 23.20–53.03 μmol L−1); ammonium (1.22 μmol L−1, 0.5–1.97 μmol L−1); phosphate (1.88 μmol L−1, 1.48–2.34 μmol L−1); and silicate (72.00 μmol L−1, 66.54–87.10 μmol L−1). The mean concentration of chl a was 0.644 μg L−1, with a range of 0.08–2.14 μg L−1.
Overall bacterial composition
A total of 129 307 sequences were obtained after quality filtering and trimming of the original sequences, which were clustered into 6195 bacterial OTUs at 97% similarity (Table 2). The mean richness of 18 water sample reads was 7183 (ranging from 5738 to 8324 reads per sample). The proportions of each bacterioplankton group, as calculated by the sequence data (Fig. 3b), showed the same trends as the OTU data (Fig. 3a), but the sequence data highlight the percentage difference among these bacterial groups (Fig. 3). For example, the most abundant order, Pelagibacterales, possessed 42.59% of the total reads but only 22.97% of the total number of OTUs.
Table 2 Estimates of coverage, phylotype richness and alpha-diversity indices for bacteria at the northern tip of the Antarctic Peninsula. | ||||||||
Label | Sample ID | Reads | OTUsa,b | ACEb | Chaob | Shannonb | Inverse Simpsonb | Coverageb |
0.03 | D1-9-2 mc | 7142 | 565 | 3978.09 | 1637.99 | 3.57 | 7.51 | 0.93 |
0.03 | D1-9-50 mc | 5738 | 601 | 5103.87 | 2203.35 | 3.38 | 5.60 | 0.92 |
0.03 | D2-3-25 md | 6369 | 643 | 4337.31 | 2299.72 | 3.58 | 7.60 | 0.92 |
0.03 | D2-5-2 md | 8252 | 459 | 3685.01 | 1569.44 | 3.02 | 5.42 | 0.94 |
0.03 | D2-5-25 md | 6149 | 710 | 10551.24 | 5313.53 | 3.96 | 12.42 | 0.90 |
0.03 | D2-8-25 me | 7105 | 533 | 3477.36 | 1503.08 | 3.23 | 7.53 | 0.93 |
0.03 | D3-1-25 md | 6534 | 653 | 4932.77 | 2263.96 | 3.43 | 6.40 | 0.92 |
0.03 | D3-3-25 mc | 7153 | 692 | 4360.22 | 1820.84 | 3.46 | 5.88 | 0.92 |
0.03 | D3-5-25 md | 6913 | 552 | 3398.94 | 1606.04 | 3.36 | 7.17 | 0.93 |
0.03 | D3-8-25 mf | 7650 | 585 | 2715.93 | 1447.65 | 3.20 | 6.72 | 0.93 |
0.03 | D4-11-25 mf | 6763 | 762 | 7682.88 | 3295.00 | 3.90 | 14.68 | 0.90 |
0.03 | D4-7-25 md | 8285 | 620 | 19283.50 | 6505.25 | 2.65 | 3.60 | 0.91 |
0.03 | D5-1-25 me | 7151 | 563 | 6031.75 | 1938.41 | 2.62 | 3.07 | 0.93 |
0.03 | D5-3-2 mc | 7780 | 634 | 5120.78 | 1728.39 | 3.17 | 5.30 | 0.92 |
0.03 | D5-3-25 md | 7531 | 658 | 12774.69 | 5341.69 | 3.13 | 5.20 | 0.91 |
0.03 | D5-6-25 me | 6439 | 600 | 4291.71 | 2340.06 | 3.03 | 4.21 | 0.92 |
0.03 | D5-8-25 mc | 8029 | 670 | 9597.99 | 4332.69 | 3.76 | 14.75 | 0.91 |
0.03 | D5-9-50 me | 8324 | 746 | 11126.81 | 7423.78 | 3.26 | 4.47 | 0.90 |
ANOVA | ||||||||
Fg | 0.875 | 0.625 | 0.675 | 2.075 | 0.764 | 0.533 | 0.871 | |
Sig.g | 0.437 | 0.549 | 0.524 | 0.160 | 0.491 | 0.598 | 0.439 | |
Fh | 0.043 | 0.428 | 0.171 | 0.132 | 1.243 | 1.416 | 0.184 | |
Sig.h | 0.987 | 0.736 | 0.914 | 0.939 | 0.331 | 0.280 | 0.906 | |
aAn OTU was defined as containing sequences with ≤3% difference. bValues are average based on rarefactions at 5738 sequences per sample. cWWs. dSSWs. eMWM. fSSWw. gResults of ANOVA were calculated on the basis of three depths: 2 m, 25 m, 50 m. hResults of ANOVA were calculated on the basis of the different water masses. | ||||||||

Alphaproteobacteria was the most abundant group, accounting for 40.71% of the total number of OTUs (55.30% of all sequences) (Fig. 3). Within this class, Pelagibacterales and Rhodobacterales were the most abundant orders, accounting for 22.97% and 14.32% of the total OTUs, respectively (Fig. 3). In total, 28 genera were identified within the class Alphaproteobacteria, with the most abundant genera being Pelagibacter (39.99% of the total sequences; exclusively Pelagibacter ubique), Octadecabacter (5.92%) and Sulfitobacter (1.82%).
Gammaproteobacteria was the second most representative bacterial class (27.31% of the total OTUs and 25.66% of all sequences) (Fig. 3) and was mainly represented by the orders Oceanospirillales (7.43% of total OTUs) and Alteromonadales (6.68% of total OTUs). Thirty-four genera were identified within the class Gammaproteobacteria. Three genera in the order Oceanospirillales were in the top 10 most relatively abundant groups, namely, Alcanivorax (1.55% of total sequences), Candidatus Portiera (1.34% of total sequences) and the SUP05 group (3.98% of total sequences). In addition, the group HTCC2207 and the genus Alteromonas of the order Alteromonadales (2.78% and 0.99% of all sequences, respectively) were found within the 10 most relatively abundant groups. The genera Glaciecola and Marinobacter of Alteromonadales (0.27% and 0.18% of all sequences) exhibited relatively high abundances, although they were not included within the top 10.
The phylum Bacteroidetes was the third most represented group with 32 genera and 93.77% of the sequences belonging to the order Flavobacteriales (Fig. 3). Within Flavobacteriales, the genera Polaribacter (more than 68% composed of Polaribacter irgensii) and Ulvibacter (with 89.19% composed of Ulvibacter litoralis) represented 7.70% and 2.00% of the total sequences, respectively.
Besides the most dominant bacterial taxa (Alphaproteobacteria, Gammaproteobacteria and Bacteroidetes), the unclassified bacterial group (3.16%), other bacteria (1.39%), other Proteobacteria (2.49%) and Cyanobacteria (1.34%) were detected with OTUs >1% (Fig. 3). Betaproteobacteria, Deltaproteobacteria, Actinobacteria, Planctomycetes and Verrucomicrobia were also detected at relative low abundances.
The most abundant bacterial order was Pelagibacterales. The highest percentage (>50%) was detected in samples collected at 25 m depth from site D4-7 (sample D4-7-25 m), while the lowest relative abundance (20%) was detected from sample D5-8-25 m. The second most abundant bacterial order was Flavobacteriales, with relative abundances varying from <10% in sample D4-7-25 m to >30% in sample D4-11-25 m. The orders Rhodobacterales and Alteromonadales presented the highest relative abundances (>20% each) in sample D5-8-25 m (Supplementary Fig. S1). The most abundant genus Pelagibacter showed the same distribution pattern as Pelagibacterales, with the highest percentage from sample D4-7-25 m and lowest percentage from sample D5-8-25 m. As for Flavobacteriales, Polaribacter exhibited the highest relative abundance from sample D4-11-25 m and the lowest from sample D4-7-25 m.
Alpha-diversity indices
The observed number of OTUs ranged from 459 to 762. Good’s coverage ranged from 90 to 94%, which indicates that > 90% of the OTUs were sequenced. Specimens from samples D4-7-25 m and D5-9-50 m exhibited higher ACE (19293.50; 11126.81) and Chao (6505.25; 7423.78) indices, which indicates higher community richness. The highest Shannon index (3.96) occurred in sample D2-5-2 m, followed by samples D4-11-25 m (3.90) and D5-8-25 m (3.76), and the highest Inverse Simpson index (14.75) was found in sample D5-8-25 m (14.75), followed by D4-11-25 m (14.68) and D3-5-2 m (12.42). The lowest values were from samples D5-1-25 m, indicating that these samples presented the highest community diversity. There was no significant difference in α-diversity indices among sampling depths or water masses (Table 2).
Correlations between environmental factors and bacterial communities
According to the pRDA results, 30.39% of the variance was explained by environmental factors. Among these variance, 13.83% was caused by nutrients (nitrite, nitrate, ammonium, phosphate, silicate) and chl a, whereas 9.42% was caused by temperature, salinity and depth. The other 7.14% could be explained by temperature, salinity, depth and nutrients (Supplementary Table S2). Axis one, which was more closely related with temperature, explained 21.57% of the data set variance, while only 4.62% of the total variance was explained by axis two, which was more closely correlated with depth, phosphate and silicate concentrations (Fig. 4).
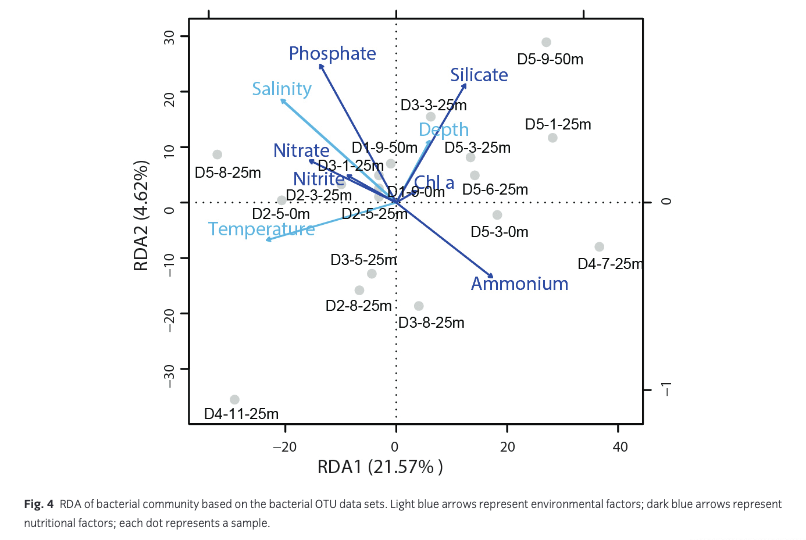
The dominant bacterial genera showed lower correlations with environmental factors (nutrients, chl a, temperature and salinity; Fig. 5, Supplementary Table S3). More than 50% of the dominant genera showed a significant correlation with only one of the measured factors. For example, Pelagibacter (r = −0.470, p = 0.049), Sulfitobacter (r = −0.513, p = 0.029) and Polaribacter (r = 0.765, p < 0.001) only showed a significant correlation with ammonium. Alteromonas (r = 0.543, p = 0.020) and Marinobacter (r = 0.649, p = 0.004) only showed significant positive correlations with nitrate. Octadecabacter, group SUP05, and Ulvibacter showed no significant correlations with any of the above factors. Two genera showed significant correlations with more than one environmental factor. Alcanivorax was positively correlated with salinity (r = 0.502, p = 0.034), phosphate (r = 0.567, p = 0.014) and nitrate (r = 0.717, p = 0.001), while Glaciecola was negatively correlated with temperature (r = −0.469, p = 0.050) and chl a (r = −0.485, p = 0.041).
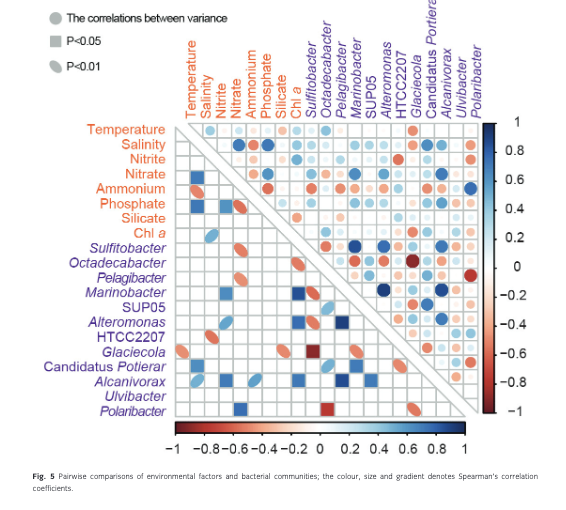
A high frequency of significant correlations was observed among interspecific populations. The genera Pelagibacter, Sulfitobacter and Glaciecola showed significant correlations with at least two genera. Pelagibacter showed positive correlations with SUP05 (r = 0.477, p = 0.045) and Candidatus Portiera (r = 0.500, p = 0.035) and a significant negative correlation with Polaribacter (r = −0.761, p < 0.001). Glaciecola showed negative correlations with group SUP05 (r = −0.497, p = 0.036) and Candidatus Portiera (r = −0.485, p = 0.041) and a significant negative correlation with Octadecabacter (r = −0.896, p < 0.001). The genus Marinobacter showed significant positive correlations with Sulfitobacter (r = 0.852, p < 0.001), Alteromonas (r = 0.911, p < 0.001) and Alcanivorax (r = 0.864, p < 0.001) and a negative correlation with Octadecabacter (r = −0.578, p < 0.012). Octadecabacter was negatively correlated with Sulfitobacter (r = −0.522, p = 0.026) and Alteromonas (r = −0.521, p = 0.027). No significant correlations were observed between the genus Ulvibacter and environmental factors, nor with other dominant bacterioplankton genera.
Discussion
Bacterioplankton community structure
The microbial communities of many Antarctic Ocean zones have been studied using molecular approaches, including the Antarctic Peninsula (Delong et al. 1994; Ducklow et al. 2012; Grzymski et al. 2012; Williams et al. 2012; Luria et al. 2014; Signori et al. 2014; Luria et al. 2016; Bowman et al. 2017; Signori et al. 2018), the Scotia Sea (Murray et al. 1998; Jamieson et al. 2012) and the Weddell Sea (Garcia-Martinez & Rodriguez-Valera 2000; Paterson et al. 2012). Less attention has been given to the bacterioplankton off the northern tip of the Antarctic Peninsula.
The most abundant bacterial group identified in the current study was Alphaproteobacteria, followed by Gammaproteobacteria and Bacteroidetes. These bacterial groups have been reported as common in other parts of the Southern Ocean in studies using similar (Ghiglione & Murray 2012) or different methods (Wilkins, Lauro et al. 2013). For example, in the eastern Scotia Sea, a similar composition was detected by fluorescence in situ hybridization; however, their composition is proportionally different from other parts of the Southern Ocean. In the western Scotia Sea (Topping et al. 2006), Gammaproteobacteria and Bacteroidetes (Sphingobacteria/Flavobacteria) exhibited lower relative abundances than in the present study. Furthermore, the dominant bacterioplankton varied among adjacent areas during different seasons. In surface waters of the Scotia Arc (Jamieson et al. 2012) and the WAP (Murray & Grzymski 2007; Signori et al. 2014), Gammaproteobacteria and CFB were the dominant community members, followed by Alphaproteobacteria. A study using length heterogeneity PCR and clone libraries showed that in early winter, Gammaproteobacteria were dominant in the north-west Weddell Sea, followed by Alphaproteobacteria and Bacteroidetes (Murray et al. 2011).
Dominant bacterioplankton have a great influence on the microenvironment that they inhabit due to their greater contribution to organic compounds cycling, such as the carbon cycle, given their adaptable metabolism. Approximately, 50% of the organic carbon is produced through heterotrophic bacterial metabolism (Ducklow 2000; Bunse & Pinhassi 2017). In addition, DOC in surface water can be reduced by bacterioplankton communities through enhanced consumption (James et al. 2017). In the current study, the most abundant bacterioplankton genera possess the ability to metabolize carbon. For example, Pelagibacter (representing 39.99% of all sequences) is effective in consuming DOC and producing nutrients that are required by algae (Giovannoni et al. 2005). Representatives of Octadecabacter are non-pigmented, psychrophilic gas-vacuolate bacteria from polar sea ice and water (Gosink et al. 1997), and Marinobacter (Alteromonadales) has the ability to biodegrade hydrophobic organic compounds, mainly in the form of lipids and hydrocarbons (Methe et al. 2005; Singer et al. 2011; Mounier et al. 2014).
The northern tip of the Antarctic Peninsula is regarded as the main gateway into the Southern Ocean and the Antarctic continent, which has been undergoing drastic changes (Meredith & King 2005; Turner et al. 2005; Parkinson & Cavalieri 2012; Meredith et al. 2014). Over the past 50 years, the sea surface temperature has increased more than 1°C (Meredith & King 2005) and the WAP glaciers have shrunk nearly 90% (Cook et al. 2016). Perennial ice is no longer the feature of this region with the average sea-ice duration shortened by 90 days (Martinson et al. 2008; Stammerjohn et al. 2012). Furthermore, DMSP concentrations have been linked to sea-ice melting, indicating that DMS production is likely to decrease with ongoing reductions in sea-ice coverage (Stefels et al. 2018). DMS production results from physiological stress such as UV radiation in low-nutrient, subtropical, oligotrophic regions (Toole & Siegel 2004). DMSP plays an important role in the global cycle of sulphur and is the major organic sulphur source in the world’s oceans. In addition, DMSP is rapidly degraded by marine bacteria that cleave DMSP to DMS. Release of DMS is a significant natural sulphur source to the atmosphere and the oxidation products of DMS cause cloud nucleation that may affect the weather and climate (Todd et al. 2007; Zeng et al. 2016). The bacterial consumption of DMS results in its loss, meanwhile DMS production by bacteria can be influenced by their need to assimilate sulphur from DMSP (Kiene & Bates 1990; Howard et al. 2006; Johnston et al. 2008; del Valle et al. 2009; Simó et al. 2009; Herrmann et al. 2012). Although no DMS data were recorded from the waters off the northern tip of Antarctic Peninsula, the data from the other Antarctica areas show that the Southern Ocean is a global hotspot for DMS production (Criscitiello et al. 2013; Kim et al. 2017; Galí et al. 2018; Stefels et al. 2018). For example, the concentrations of DMS in the surface water of the Amundsen Sea Polynya and Pine Island Polynya is up to 400 nM (Kim et al. 2017). In the current study, several abundant genera possess the ability to transform inorganic and organic sulphur compounds (Gonzalez et al. 1999; Dickinson et al. 2016; Zeng et al. 2016; Roseobase genomic database for marine Roseobacters: http://www.roseobase.org/Species/sulf_nas14_1.html). The most studied genera are Sulfitobacter (Rhodobacterales), which metabolizes taurine and organosulphur compounds such as DMSP and releases DMS and CO2 into the environment (Amin et al. 2015). Furthermore, the strain Sulfitobacter sp. NAS-14.1 uses DMSP as a sole carbon source (Dickinson et al. 2016). The group HTCC2207 (Gammaproteobacteria) harbours the ddd (DMSP-dependent DMS) genes that are involved in the production of DMS (Lovelock et al. 1972).
Impact of environment
Although previous data show that the bacterioplankton community is dynamic and influenced by both location and season (sea-ice melting, ice-island run off) (Moline et al. 2009; Montes-Hugo et al. 2009; Signori et al. 2018), the current study indicates that nutrients plus chl a contribute more to bacterioplankton diversity and community structure than water masses and depth (Fig. 5, Supplementary Fig. S1, Supplementary Table S2). The environmental factors had a limited influence on the bacterial community structure. According to the RDA and pRDA results, only 30.39% of the variation could be explained by environmental factors (Supplementary Table S2). Furthermore, no significant difference in nutrients and chl a concentration was observed among water masses or sampling depth, as revealed by ANOVA (Supplementary Table S4). This indicates that the nutrients and chl a of the surface waters at the northern tip of the Antarctic Peninsula become homogeneous in summer, when the water masses are mixed.
Sea ice and glacier retreat together with wind could influence the concentration of nutrients and chl a by reducing the differences among water masses. Sea-ice and glacier melting are important physical determinants of spatial and temporal changes in the structure of microbial communities (Dierssen et al. 2002). Meltwater-related stratification and the transition to non-stabilized Antarctic surface waters have impacts on both micro-eukaryotic and bacterial community compositions (Piquet et al. 2011). The northern tip of the Antarctic Peninsula is located at the Antarctic Polar Front, where small temperature changes can have large effects on the extent and thickness of the sea ice (Lovelock et al. 1972; Smetacek & Nicol 2005) and where glacier ice has retreated in recent decades (Wouters et al. 2015). Besides sea-ice and glacier meltwater, wind also affects the streaming of Antarctic surface water, speeding the confluence of different water masses. As a result, wind promotes a mixing of the bacteria in the north-western Antarctic Peninsula, influencing the community down to a depth of ca. 100 m (Signori et al. 2014). Sea-ice and glacier meltwater, along with wind, may explain why the dominant bacteria did not vary with water mass.
Biological interactions
Biological interactions, such as with phytoplankton and zooplankton, determine bacterial mortality rates. These interactions also have the potential to induce differential effects on bacterial populations (Säwström et al. 2009; Hitchcock et al. 2010; Dang & Lovell 2016; Luria et al. 2016; Bunse & Pinhassi 2017; Luria et al. 2017). Phytoplankton-derived organic matter appears to trigger the observed shifts in microbial communities, with higher relative abundance of Rhodobacteraceae than Pelagibacteraceae in January, when phytoplankton blooms often occur (Luria et al. 2016; Luria et al. 2017). However, our samples were likely taken at the beginning of a return to a pre-bloom state, which usually occurs during late January or early February in the WAP (Luria et al. 2016).
Spearman analysis shows that 10 out of the 12 dominant bacterial genera were significantly correlated with the presence of at least two other genera (Supplementary Table S3). In some cases, no correlation was found between the physicochemical parameters and bacterioplankton distribution (Zubkov et al. 2002), similar to Octadecabacter and the group SUP05 in the present investigation. The diversity, biomass and community structure of bacterioplankton are affected not only by environmental factors such as temperature, salinity, depth and nutrients (Church et al. 2003; Gilbert et al. 2012), and the presence of eukaryotes (planktonic algae; Limardo & Worden 2015), but also by the bacterial community members themselves, especially in surface waters (Dang & Lovell 2016).
Our data also suggest that both cooperation and competition happen among the dominant bacterioplankton groups. For example, the positive correlation between Sulfitobacter and Marinobacter, Alteromonas and Alcanivorax would suggest cooperative relationships, as the latter group are able to utilize hydrocarbons, whereas Sulfitobacter is a sulphite-oxidizing bacterium that lacks aerobic anoxygenic photosynthesis abilities (Kostka et al. 2011; Math et al. 2012; Mas-Lladó et al. 2014). A cooperative relationship would be beneficial for the distribution of these groups. Some bacterial groups are strongly associated with particular ecological niches. This includes members of the order Pelagibacterales (class Alphaproteobacteria), which are key colonizers of surface marine waters (Wilkins, Yau et al. 2013; Dang & Lovell 2016). In contrast, a competitive relationship would benefit bacterial survival in areas with limited resources. This is demonstrated by the reported overlap in ecological function concerning DMSP degradation by Octadecabacter and Sulfitobacter (Lovelock et al. 1972; Wilkins, Yau et al. 2013), which reflects the negative correlation of these genera detected in the current study. Conversely, with respect to the utilization of organic carbon, Octadecabacter, Marinobacter, Alteromonas and Glaciecola were reported to have similar ecological functions (Methe et al. 2005; Singer et al. 2011; Wilkins, Yau et al. 2013; Mounier et al. 2014); however, negative correlations between them were also detected here.
Conclusions
Significant differences were not found between the nutrients and chl a concentrations, and water masses or sampling depth. These results suggest that the surface waters off the northern tip of the Antarctic Peninsula are homogeneous during the summer. Environmental factors measured in the current study contribute relatively little to the variation in bacterioplankton diversity and community structure. More significant correlations were found within the dominant genera than between the bacterial groups and the environmental factors, suggesting that environmental parameters are less important than biological interactions for influencing bacterioplankton diversity and community structure in the waters by the northern tip of the Antarctic Peninsula.
Availability of data and materials
The data sets generated and analysed during the current study have been submitted to the National Centre for Biotechnology Information Sequence Read Archive (SRA: SRP077881) under BioProject ID PRJNA299749. An MIMS file of the northern tip of the Antarctic Peninsula is provided as Supplementary Table S5.
Acknowledgements
We are grateful to Professor Shi Jiuxin of Ocean University of China for kindly providing temperature and salinity data and to Professor Pan Jianming of the Second Institute of Oceanography for providing nutrient data. The cruise was supported by the RV Xuelong ice-breaker.
References
- Agogué H., Lamy D., Neal P.R., Sogin M.L. & Herndl G.J. 2011. Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Molecular Ecology 20, 258–274, http://dx.doi.org/10.1111/j.1365-294X.2010.04932.x.
- Amin S.A., Hmelo L.R., van Tol H.M., Durham B.P., Carlson L.T., Heal K.R., Morales R.L., Berthinume C.T., Parker M.S., Djunaedi B., Ingalls A.E., Parsek M.R., Moran M.A. & Armbrust E.V. 2015. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101, http://dx.doi.org/10.1038/nature14488.
- Anisimov O.A., Vaughan D.G., Callaghan T.V., Furgal C.M., Marchant H., Prowse T.D. & Walsh J.E. 2007. Polar regions (Arctic and Antarctic). In M.L. Parry et al. (eds.): Climate change 2007: impacts, adaptation and vulnerability. Contribution of Working Group II to the fourth assessment report of the Intergovernmental Panel on Climate Change. Pp. 653–685. Cambridge: Cambridge University Press.
- Azam F. 1998. Microbial control of oceanic carbon flux: the plot thickens. Science 280, 694–696, http://dx.doi.org/10.1126/science.280.5364.694.
- Azam F. & Malfatti F. 2007. Microbial structuring of marine ecosystems. Nature Review Microbiology 5, 782–791, http://dx.doi.org/10.1038/nrmicro1747.
- Bowman J.S., Amaral-Zettler L.A., Rich J.J., Luria C.M. & Ducklow H.W. 2017. Bacterial community segmentation facilitates the prediction of ecosystem function along the coast of the western Antarctic Peninsula. The ISME Journal 11, 1460–1471, http://dx.doi.org/10.1038/ismej.2016.204.
- Bunse C. & Pinhassi J. 2017. Marine bacterioplankton seasonal succession dynamics. Trends in Microbiology 25, 494–505, http://dx.doi.org/10.1016/j.tim.2016.12.013.
- Church M.J., Delong E.F., Ducklow H.W., Karner M.B., Preston C.M. & Karl D.M. 2003. Abundance and distribution of planktonic archaea and bacteria in the waters west of the Antarctic Peninsula. Limnology and Oceanography 48, 1893–1902, http://dx.doi.org/10.4319/lo.2003.48.5.1893.
- Cook A.J., Holland P.R., Meredith M.P., Murray T., Luckman A. & Vaughan D.G. 2016. Ocean forcing of glacier retreat in the western Antarctic Peninsula. Science 353, 283–286, http://dx.doi.org/10.1126/science.aae0017.
- Cook A.J. & Vaughan D.G. 2010. Overview of areal changes of the ice shelves on the Antarctic Peninsula over the past 50 years. Cryosphere 4, 77–98, http://dx.doi.org/10.5194/tc-4-77-2010.
- Criscitiello A.S., Das S.B., Evans M.J., Frey K.E., Conway H., Joughin I., Medley B. & Steig E.J. 2013. Ice sheet record of recent sea-ice behavior and polynya variability in the Amundsen Sea, West Antarctica. Journal of Geophysical Research—Oceans 118, 118–130, http://dx.doi.org/10.1029/2012JC008077.
- Dang H.Y. & Lovell C.R. 2016. Microbial surface colonization and biofilm development in marine environments. Microbiology Molecular Biology Review 80, 91–138, http://dx.doi.org/10.1128/MMBR.00037-15.
- Delille D. 2004. Abundance and function of bacteria in the Southern Ocean. Cell Molecular Biology 50, 543–551.
- Delong E.F., Wu K.Y., Prezelin B.B. & Jovine R.V. 1994. High abundance of archaea in Antarctic marine picoplankton. Nature 371, 695–697, http://dx.doi.org/10.1038/371695a0.
- del Valle D.A., Kieber D.J., Toole D.A., Brinkley J. & Kiene R.P. 2009. Biological consumption of dimethylsulfide (DMS) and its importance in DMS dynamics in the Ross Sea, Antarctica. Limnology and Oceanography 54, 785–798.
- Dickinson I., Goodall-Copestake W., Thorne M.A.S., Schlitt T., Ávila-Jiménez M.L. & Pearce D.A. 2016. Extremophiles in an Antarctic marine ecosystem. Microorganisms 4(1), UNSP 8, http://dx.doi.org/10.3390/microorganisms4010008.
- Dierssen H.M., Smith R.C. & Vernet M. 2002. Glacial meltwater dynamics in coastal waters west of the Antarctic Peninsula. Proceedings of the National Academy of Sciences of the United States of America 99, 1790–1795, http://dx.doi.org/10.1073/pnas.032206999.
- Doney S.C., Ruckelshaus M., Duffy J.E., Barry J.P., Chan F., English C.A., Galindo H.M., Grebmeier J.M., Hollowed A.B., Knowlton N., Polovina J., Rabalais N.N. Sydeman W.J. & Talley L.D. 2012. Climate change impacts on marine ecosystems. Annual Review on Marine Science 4, 11–37, http://dx.doi.org/10.1146/annurev-marine-041911-111611.
- Ducklow H. 1999. The bacterial component of the oceanic euphotic zone. FEMS Microbiol Ecology 30, 1–10, http://dx.doi.org/10.1111/j.1574-6941.1999.tb00630.x.
- Ducklow H.W. 2000. Bacterial production and biomass in the oceans. In D.L. Kirchman (ed.): Microbial ecology of the oceans. Pp. 85–120. New York: John Wiley & Sons.
- Ducklow H.W., Fraser W.R., Meredith M.P., Stammerjohn S.E., Doney S.C., Martinson D.G., Sailley S.F., Schofield O.M., Steinberg D.K., Venables H.J. & Amsler C.D. 2013. West Antarctic Peninsula: an ice-dependent coastal marine ecosystem in transition. Oceanography 26, 190–203, http://dx.doi.org/10.5670/oceanog.2013.62.
- Galí M., Levasseur M., Devred E., Simó R. & Babin M. 2018. Sea-surface dimethylsulfide (DMS) concentration from satellite data at global and regional scales. Biogeosciences 15, 3497–3519, http://dx.doi.org/10.5194/bg-15-3497-2018.
- Garcia-Martinez J. & Rodriguez-Valera F. 2000. Microdiversity of uncultured marine prokaryotes: the SAR11 cluster and the marine Archaea of Group I. Molecular Ecology 9, 935–948, http://dx.doi.org/10.1046/j.1365-294x.2000.00953.x.
- Ghiglione J.F. & Murray A.E. 2012. Pronounced summer to winter differences and higher wintertime richness in coastal Antarctic marine bacterioplankton. Environmental Microbiology 14, 617–629, http://dx.doi.org/10.1046/10.1111/j.1462-2920.2011.02601.x.
- Gilbert J.A., Steele J.A., Caporaso J.G., Steinbruck L., Reeder J., Temperton B., Huse S., McHardy A.C., Knight R., Joint I., Somerfield P., Fuhrman J.A. & Field D. 2012. Defining seasonal marine microbial community dynamics. The ISME Journal 6, 298–308, http://dx.doi.org/10.1038/ismej.2011.107.
- Giovannoni S.J., Tripp H.J., Givan S., Podar M., Vergin K.L., Baptista D., Bibbs L., Eads J., Richardson T.H., Noordewier M., Rappe M.S., Short J.M., Carrington J.C. & Mathur E.J. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245, http://dx.doi.org/10.1126/science.1114057.
- Gonzalez J.M., Kiene R.P. & Moran M.A. 1999. Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Applied and Environmental Microbiology 65, 3810–3819.
- Gordon A.L. 1967. Structure of Antarctic waters between 20°W and 170°W. Antarctic Map Folio Series, Number 6. New York: American Geographical Society.
- Gordon A.L. & Huber B.A. 1995. Warm Weddell Deep Water west of Maud Rise. Journal of Geophysical Research—Oceans 100, 13747–13753, http://dx.doi.org/10.1029/95JC01361.
- Gosink J.J., Herwig R.P. & Staley J.T. 1997. Octadecabacter arcticus gen. nov., sp. nov., and O. antarcticus, sp. nov., nonpigmented, psychrophilic gas vacuolate bacteria from polar sea ice and water. Systematic Applied Microbiology 20, 356–365, http://dx.doi.org/10.1016/S0723-2020(97)80003-3.
- Grasshoff K., Kremling K. & Ehrhardt M. 2007. Methods of seawater analysis. Weinheim, Germany: Wiley-VCH Verlag GmbH.
- Grzymski J.J., Riesenfeld C.S., Williams T.J., Dussaq A.M., Ducklow H. Erickson M, Cavicchioli R. & Murray A.E. 2012. A metagenomic assessment of winter and summer bacterioplankton from Antarctica Peninsula coastal surface waters. The ISME Journal 6, 1901–1915, http://dx.doi.org/10.1038/ismej.2012.31.
- He J.F., Zhang F., Lin L., Ma Y.X. & Chen J.F. 2012. Bacterioplankton and picophytoplankton abundance, biomass, and distribution in the western Canada Basin during summer 2008. Deep-Sea Research Part II 81–84, 36–45, http://dx.doi.org/10.1016/j.dsr2.2012.08.018.
- Herrmann M., Najjara R.G., Neeley A.R., Vila-Costa M., Dacey J.W.H., DiTullio G.R., Kieber D.J., Kiene R.P., Matrai P.A., Simó R. & Vernet M. 2012. Diagnostic modeling of dimethylsulfide production in coastal water west of the Antarctic Peninsula. Continental Shelf Research 32, 96–109.
- Hitchcock J.N., Mitrovic S.M., Kobayashi T. & Westhorpe D. 2010. Responses of estuarine bacterioplankton, phytoplankton and zooplankton to dissolved organic carbon (DOC) and inorganic nutrient additions. Estuaries and Coasts 33, 78–91, http://dx.doi.org/10.1007/s12237-009-9229-x.
- Howard E.C., Henriksen J.R., Buchan A., Reisch C.R., Bürgmann H., Welsh R., Ye W., González J.M., Mace K., Joye S.B., Kiene R.P., Whitman W.B. & Moran M.A. 2006. Bacterial taxa that limit sulfur flux from the ocean. Science 314, 649–652, http://dx.doi.org/10.1126/science.1130657.
- James A.K., Passow U., Brzezinski M.A., Parsons R.J., Trapani J.N. & Carlson C.A. 2017. Elevated pCO2 enhances bacterioplankton removal of organic carbon. PLoS One 12, e0173145, http://dx.doi.org/10.1371/journal.pone.0173145.
- Jamieson R.E., Rogers A.D., Billett D.S., Dan A.S. & Pearce D.A. 2012. Patterns of marine bacterioplankton biodiversity in the surface waters of the Scotia Arc, Southern Ocean. FEMS Microbiology Ecology 80, 452–468, http://dx.doi.org/10.1111/j.1574-6941.2012.01313.x.
- Johnston A.W., Todd J.D., Sun L., Nikolaidou-Katsaridou M.N., Curson A.R. & Rogers R. 2008. Molecular diversity of bacterial production of the climate-changing gas, dimethyl sulphide, a molecule that impinges on local and global symbioses. Journal of Experimental Botany 59, 1059–1067, http://dx.doi.org/10.1093/jxb/erm264.
- Kiene R.P. & Bates T.S. 1990. Biological removal of dimethyl sulfide from sea water. Nature 345, 702–704, http://dx.doi.org/10.1038/345702a0.
- Kim I., Hahm D., Park K., Lee Y., Choi J., Zhang M., Chen L., Kime H. & Lee S.H. 2017. Characteristics of the horizontal and vertical distributions of dimethyl sulfide throughout the Amundsen Sea Polynya. Science of the Total Environment 584–585, 154–163, http://dx.doi.org/10.1016/j.scitotenv.2017.01.165.
- Kirchman D.L., Moran X.A.G. & Kucklow H. 2009. Microbial growth in the polar oceans—role of temperature and potential impact of climate change. Nature Reviews Microbiology 7, 451–459.
- Kostka J.E., Prakash O., Overholt W.A., Green S.J., Freyer G., Canion A., Delgardio J., Norton N., Hazen T.C. & Huettel M. 2011. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico Beach sands impacted by the deepwater horizon oil spill. Applied & Environment Microbiology 77, 7962–7974, http://dx.doi.org/10.1038/nrmicro2115.
- Lepš S. & Šmilauer P. 2003. Multivariate analysis of ecological data using CANOCO. Cambridge: Cambridge University Press.
- Limardo A.J. & Worden A.Z. 2015. Microbiology: exclusive networks in the sea. Nature 522, 36–37 http://dx.doi.org/10.1038/nature14530.
- Loeb V., Siegel V., Holm-Hansen O., Hewitt R.P., Fraser W.R. & Trivelpiece W.Z. 1997. Effects of sea-ice extent and krill or salp dominance on the Antarctic food web. Nature 387, 897–900, http://dx.doi.org/10.1038/43174.
- Lovelock J.E., Maggs R.J. & Rasmussen R.A. 1972. Atmospheric dimethyl sulphide and the natural sulphur cycle. Nature 237, 452–453.
- Luria C.M., Amaral-Zettler L.A., Ducklow H.W., Repeta D.J., Rhyne A.L. & Rich J.J. 2017. Seasonal shifts in bacterial community responses to phytoplankton-derived dissolved organic matter in the western Antarctic Peninsula. Frontiers in Microbiology 8, article no. 2177, http://dx.doi.org/10.3389/fmicb.2017.02117.
- Luria C.M., Amaral-Zettleer L.A., Ducklow H.W. & Rich J.J. 2016. Seasonal succession of free-living bacterial communities in coastal waters of the western Antarctic Peninsula. Frontiers in Microbiology 7, article no. 1731, http://dx.doi.org/10.3389/fmicb.2016.01731.
- Luria C.M., Ducklow H.W. & Amaral-Zettler L.A. 2014. Marine bacterial, archaeal and eukaryotic diversity and community structure on the continental shelf of the western Antarctic Peninsula. Aquatic Microbiology Ecology 73, 107–121, http://dx.doi.org/10.3354/ame01703.
- Manganelli M., Malfatti F., Samo T.J., Mitchell B.G., Wang H. & Azam F. 2009. Major role of microbes in carbon fluxes during austral winter in the southern Drake Passage. PLoS One 4, e6941, http://dx.doi.org/10.1371/journal.pone.0006941.
- Martinson D.G., Stammerjohn S.E., Iannuzzi R.A., Smith R.C. & Vernet M. 2008. Western Antarctic Peninsula physical oceanography and spatio-temporal variability. Deep-Sea Research Part II 55, 1964–1987, http://dx.doi.org/10.1016/j.dsr2.2008.04.038.
- Mas-Lladó M., Piña-Villalonga J.M., Brunet-Galmés I., Nogales B. & Bosch R. 2014. Draft genome sequences of two isolates of the Roseobacter group, Sulfitobacter sp. strains 3SOLIMAR09 and 1FIGIMAR09, from harbors of Mallorca Island (Mediterranean Sea). Genome Announcement 2, e00350-14, http://dx.doi.org/10.1128/genomeA.00350-14.
- Math R.K., Jin H.M., Kim J.M., Hahn Y., Park W., Madsen E.L. & Jeon C.O. 2012. Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS One 7, e35784, http://dx.doi.org/10.1371/journal.pone.0035784.
- McDonald D., Price M.N., Goodrich J., Nawrocki E.P., DeSantis T.Z., Probst A., Andersen G.L., Knight R. & Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME Journal 6, 610–618, http://dx.doi.org/10.1038/ismej.2011.139.
- Meredith M.P., Jullion L., Brown P.J., Naveira-Garabato A.C. & Couldrey M.P. 2014. Dense waters of the Weddell and Scotia seas: recent changes in properties and circulation. Philosophical Transactions A 372, article no. 20130041, http://dx.doi.org/10.1098/rsta.2013.0041.
- Meredith M.P. & King J.C. 2005. Rapid climate change in the ocean west of the Antarctic Peninsula during the second half of the 20th century. Geophysical Research Letters 32, L19604, http://dx.doi.org/10.1029/2005GL024042.
- Methe B.A., Nelson K.E., Deming J.W., Momen B., Melamud E., Zhang X., Moult J., Madupu R., Nelson W.C., Dodson R.J., Leone L.B., Daugherty S.C., Durkin A.S., DeBoy R.T., Kolonay J.F., Sullivan S.A., Zhou L., Davidsen T.M., Wu M., Huston A.L., Lewis M., Weaver B., Weidman J.F., Khouri H., Utterback T.R., Feldblyum T.V. & Fraser C.M. 2005. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proceedings of the National Academy of Sciences of the United States of America 102, 10913–10918, http://dx.doi.org/10.1073/pnas.0504766102.
- Moline M.A., Claustre H., Frazer T.K., Schofield O. & Vernet M. 2004. Alteration of the food web along the Antarctic Peninsula in response to a regional warming trend. Global Change Biology 10, 1973–1980, http://dx.doi.org/10.1111/j.1365-2486.2004.00825.x.
- Moline M.A., Karnovsky N.J., Brown Z., Divoky G.J., Frazer T.K., Jacoby C.A., Torres J.J. & Fraser W.R. 2009. High latitude changes in ice dynamics and their impact on polar marine ecosystems. Annals of the New York Academy of Sciences 1134, 267–319, http://dx.doi.org/10.1196/annals.1439.010.
- Montes-Hugo M., Doney S.C., Ducklow H.W., Fraser W., Martinson D., Stammerjohn S.E. & Schofield O. 2009. Recent changes in phytoplankton communities associated with rapid regional climate change along the western Antarctic Peninsula. Science 323, 1470–1473, http://dx.doi.org/10.1126/science.1164533.
- Mounier J., Camus A., Mitteau I., Vaysse P.J., Goulas P., Grimaud R. & Sivadon P. 2014. The marine bacterium Marinobacter hydrocarbonoclasticus SP17 degrades a wide range of lipids and hydrocarbons through the formation of oleolytic biofilms with distinct gene expression profiles. FEMS Microbiology Ecology 90, 816–831, http://dx.doi.org/10.1111/1574-6941.12439.
- Murray A.E. & Grzymski J. 2007. Diversity and genomics of Antarctic marine micro-organisms. Philosophical Transactions of the Royal Society B 362, 2259–2271, http://dx.doi.org/10.1098/rstb.2006.1944.
- Murray A.E., Peng V., Tyler C. & Wagh P. 2011. Marine bacterioplankton biomass, activity and community structure in the vicinity of Antarctic icebergs. Deep-Sea Research Part II 58, 1407–1421, http://dx.doi.org/10.1016/j.dsr2.2010.11.021.
- Murray A.E., Perston C.M., Massana R., Taylor L.T., Blakis A., Wu K. & Delong E.F. 1998. Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Applied Environmental Microbiology 64, 2585–2595.
- Oksanen J., Blanchet F.G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpson G.L., Solymos P., Stevens M.H.H., Szoecs E. & Wagner H. 2018. Vegan: community ecology package, version. R package version 2.4.6. Accessed on the internet at http://CRAN.R-project.org/package=vegan on 20 May 2018.
- Parkinson C.L. & Cavalieri D.J. 2012. Antarctic sea ice variability and trends, 1979–2010. Cryosphere 6, 871–880, http://dx.doi.org/10.5194/tc-6-871-2012.
- Parsons T.R. 1984. A manual of chemical & biological methods for seawater analysis. Amsterdam: Pergamon.
- Paterson H., Laybourn-Parry J., Paterson H. & Laybourn-Parry J. 2012. Antarctic sea ice viral dynamics over an annual cycle. Polar Biology 35, 491–497.
- Piquet A.M., Bolhuis H., Meredith M.P. & Buma A.G. 2011. Shifts in coastal Antarctic marine microbial communities during and after melt water-related surface stratification. FEMS Microbiology Ecology 76, 413–427, http://dx.doi.org/10.1111/j.1574-6941.2011.01062.x.
- Pruesse E., Quast C., Knittel K., Fuchs B.M., Ludwig W., Peplies J. & Glockner F.O. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research 35, 7188–7196, http://dx.doi.org/10.1093/nar/gkm864.
- Säwström C., Karlsson J., Laybourn-Parry J. & Granéli W. 2009. Zooplankton feeding on algae and bacteria under ice in Lake Druzhby, East Antarctica. Polar Biology 32, 1195–1202, http://dx.doi.org/10.1007/s00300-009-0619-0.
- Schlitzer R. 2018. Ocean data view, version 4.5.0. Accessed on the internet at http://odv.awi.de/ on 1 May 2018.
- Schloss P.D., Westcott S.L., Ryabin T., Hall J.R., Hartmann M., Hollister E.B., Lesniewski R.A., Oakley B.B., Parks D.H., Robinson C.J., Sahl J.W., Stres B., Thallinger G.G., Van Horn D.J. & Weber C.F. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75, 7537–7541, http://dx.doi.org/10.1128/AEM.01541-09.
- Schofield O., Ducklow H.W., Martinson D.G., Meredith M.P., Moline M.A. & Fraser W.R. 2010. How do polar marine ecosystems respond to rapid climate change? Science 328, 1520–1523, http://dx.doi.org/10.1126/science.1185779.
- Shi J.X., Sun Y.M., Jiao Y.T., Hao G. & Wang M. 2016. Water masses and exchanges in the region around the northern tip of Antarctic Peninsula observed in summer of 2011/2012. Advance in Polar Science 28, 67–79.
- Signori C.N., Pellizari V.H., Enrich-Prast A. & Sievert S.M. 2018. Spatiotemporal dynamics of marine bacterial and archaeal communities in surface waters off the northern Antarctic Peninsula. Deep-Sea Research Part II 149, 150–160, http://dx.doi.org/10.1016/j.dsr2.2017.12.017.
- Signori C.N., Thomas F., Enrich-Prast A., Pollery R.C.G. & Sievert S.M. 2014. Microbial diversity and community structure across environmental gradients in Bransfield Strait, western Antarctic Peninsula. Frontiers in Microbiology 5, article no. 647, http://dx.doi.org/10.3389/fmicb.2014.00647.
- Simó R., Vila-Costa M., Alonso-Sáez L., Cardelús C., Guadayol O., Vázquez-Domínguez E., Gasol J.M. 2009. Annual DMSP contribution to S and C fluxes through phytoplankton and bacterioplankton in a NW Mediterranean coastal site. Aquatic Microbialogy Ecology 57, 43–55, http://dx.doi.org/10.3354/ame01325.
- Singer E., Webb E.A., Nelson W.C., Heidelberg J.F., Ivanova N., Pati A. & Edwards K.J. 2011. Genomic potential of Marinobacter aquaeolei, a biogeochemical “opportunitroph”. Applied and Environmental Microbiology 77, 2763–2771, http://dx.doi.org/10.1128/AEM.01866-10.
- Smetacek V. & Nicol S. 2005. Polar ocean ecosystems in a changing world. Nature 437, 362–368, http://dx.doi.org/10.1038/nature04161.
- Stammerjohn S., Massom R., Rind D. & Martinson D. 2012. Regions of rapid sea ice change: an inter-hemispheric seasonal comparison. Geophysical Research Letters 39, L06501, http://dx.doi.org/10.1029/2012GL050874.
- Stefels J., van Leeuwe M.A., Jones E.M., Meredith M.P., Venables H.J., Webb A.L., Sian F. & Henley S.F. 2018. Impact of sea-ice melt on dimethyl sulfide (sulfoniopropionate) inventories in surface waters of Marguerite Bay, west Antarctic Peninsula. Philosophical Transactions A 376, article no. 20170169, http://dx.doi.org/10.1098/rsta.2017.0169.
- Sun F.L., Wang Y.S., Wu M.L., Jiang Z.Y., Sun C.C. & Chen H. 2014. Genetic diversity of bacterial communities and gene transfer agents in northern South China Sea. PLoS One 9, e111892, http://dx.doi.org/10.1371/journal.pone.0111892.
- Todd J.D., Rogers R., Li Y.G., Wexler M., Bond P.L., Sun L., Curson A.R.J., Malin G., Steinke M. & Johnston A.W.B. 2007. Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315, 666–669, http://dx.doi.org/10.1126/science.1135370.
- Toole D.A. & Siegel D.A. 2004. Light-driven cycling of dimethylsulfide (DMS) in the Sargasso Sea: closing the loop. Geophysical Research Letters 31, L09308, http://dx.doi.org/10.1029/2004GL019581.
- Topping J.N., Heywood J.L., Ward P. & Zubkov M.V. 2006. Bacterioplankton composition in the Scotia Sea, Antarctica, during the austral summer of 2003. Aquatic Microbiology Ecology 45, 229–235, http://dx.doi.org/10.3354/ame045229.
- Turner J., Colwell S.R., Marshall G.J., Lachlan-Cope T.A., Carleton A.M., Jones P.D. & Iagovkina S. 2005. Antarctic climate change during the last 50 years. International Journal of Climatology 25, 279–294, http://dx.doi.org/10.1002/joc.1130.
- Turner S., Pryer K.M., Miao V.P. & Palmer J.D. 1999. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. Journal of Eukaryotic Microbiology 47, 327–338, http://dx.doi.org/10.1111/j.1550-7408.1999.tb04612.x.
- Wilkins D., Lauro F.M., Williams T.J., Demaere M.Z., Brown M.V., Hoffman J.M., Andrews-Pfannkoch C., Mcquaid J.B., Riddle M.J., Rintoul S.R. & Cavicchioli R. 2013. Biogeographic partitioning of Southern Ocean microorganisms revealed by metagenomics. Environmental Microbiology 15, 1318–1333, http://dx.doi.org/10.1111/1462-2920.12035.
- Wilkins D., Yau S., Williams T.J., Allen M.A., Brown M.V., MeMaere M.Z., Lauro F.M. & Cavicchioli R. 2013. Key microbial drivers in Antarctic aquatic environments. FEMS Microbiology Review 37, 303–335, http://dx.doi.org/10.1111/1574-6976.12007.
- Williams T.J., Long E., Evans F., Demaere M.Z., Lauro F.M., Raftery M.J., Ducklow H., Grzymski J.J., Murray A.E. & Cavicchioli R. 2012. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. The ISME Journal 6, 1883–1900, http://dx.doi.org/10.1038/ismej.2012.28.
- Wouters B., Martin-Espanol A., Helm V., Flament T., van Wessem J.M., Ligtenberg S.R. & van den Broke M.R. 2015. Dynamic thinning of glaciers on the Southern Antarctic Peninsula. Science 348, 899–903, http://dx.doi.org/10.1126/science.aaa5727.
- Zeng Y.X., Qiao Z.Y., Yu Y., Li H.R. & Luo W. 2016. Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Scientific Report 6, article no. 33031, http://dx.doi.org/10.1038/srep33031.
- Zhang F., He J.F., Lin L. & Jin H.Y. 2015. Dominance of picophytoplankton in the newly open surface water of the central Arctic Ocean. Polar Biology 38, 1081–1089, http://dx.doi.org/10.1007/s00300-015-1662-7.
- Zubkov M.V., Fuchs B.M., Tarran G.A., Burkill P.H. & Amann R. 2002. Mesoscale distribution of dominant bacterioplankton groups in the northern North Sea in early summer. Aquatic Microbiology Ecology 29, 135–144, http://dx.doi.org/10.3354/ame029135.
Похожие статьи
Agnieszka Promińska
Waldemar Walczowski
Eva Falck