Эта статья опубликована под лицензией Creative Commons и не автором статьи. Поэтому если вы найдете какие-либо неточности, вы можете исправить их, обновив статью.

Инсулинорезистентность в патогенезе сахарного диабета 2 типа



Александр Юрьевич Майоров
Опубликована Янв. 1, 2011
Последнее обновление статьи Ноя. 23, 2022
Эта статья опубликована под лицензией
")



Аннотация
В статье дается обзор механизмов нарушения чувствительности к инсулину в процессе эволюции нарушений углеводного обмена: от на-рушенной гликемии натощак (НГН) до нарушенной толерантности к глюкозе (НТГ) и сахарного диабета 2 типа (СД2). Рассмотренырецепторные и пострецепторные нарушения утилизации глюкозы тканями, влияние на инсулинорезистентность (ИР) таких факторов,как глюкозотоксичность и липотоксичность. Приведены собственные данные изучения ИР у больных с различными нарушениями углевод-ного обмена. Было отмечено более выраженное снижение чувствительности к инсулину при СД2, чем при НТГ и НГН (в среднем на 50, 25и 15% соответственно по сравнению со здоровыми лицами). Продемонстрированы достоверные корреляции М-индекса с индексом массытела (ИМТ), уровнем метаболического контроля (HbA1c, триглицериды). Проанализированы различия в клинических и биохимическихпоказателях у больных СД2 в зависимости от степени ИР. При исследовании ряда гормонов и цитокинов было выявлено, что уровниадипонектина и резистина у больных СД2 были ниже по сравнению со здоровыми лицами, а уровни фактора некроза опухолей-альфаи проинсулина - выше. Приведены данные по улучшению чувствительности к инсулину на фоне лечения метформином, пиоглитазоном,инсулином, в том числе у больных с ранними нарушениями углеводного обмена. Результаты исследования говорят о необходимости ин-тенсификации сахароснижающей терапии, не дожидаясь выраженного ухудшения чувствительности к инсулину.
Ключевые слова
Инсулинорезистентность, сахарный диабет 2 типа, нарушенная толерантность к глюкозе, нарушенная гликемия натощак, М-индекс, утилизация глюкозы тканями
Впервые Himsworth и Kerr использовали термин нечувствительности к инсулину (синоним ИР) для определения относительно плохого ответа на введение экзогенного инсулина у больных сахарным диабетом (СД) и ожирением [1]. В широком смысле слова под ИР понимают снижение биологического ответа к одному или нескольким эффектам действия инсулина. Однако более часто ИР определяют как состояние, которое сопровождается снижением утилизации глюкозы тканями (УГТ) организма под влиянием инсулина, т.е. резистентностью клеток различных органов и тканей к сахароснижающему действию инсулина [2, 3]. Но поскольку биологическое действие инсулина заключается в регуляции метаболических реакций (обмен углеводов, жиров и белков) и митогенных процессов (процессов роста, дифференцировки тканей, синтеза ДНК, транскрипции генов), современное понятие ИР не сводится к параметрам, характеризующим только метаболизм углеводов, а включает также изменения метаболизма жиров, белков, функции клеток эндотелия, экспрессии генов и др. [3–6].
Наряду с термином «инсулинорезистентность» существует концепция «синдрома инсулинорезистентности» (метаболического синдрома). Он представляет собой сочетание клинических и лабораторных проявлений: нарушение углеводного обмена: нарушение гликемии натощак (НГН), нарушение толерантности к глюкозе (НТГ) или СД, центральное ожирение, дислипидемия (повышение уровня триглицеридов и ЛПНП, снижение ЛПВП), артериальная гипертония, увеличение уровня тромботических и антифибринолитических факторов и, в конечном итоге, высокая предрасположенность к развитию атеросклероза и сердечно-сосудистых заболеваний [7, 8].
ИР является центральным механизмом эволюции СД 2 типа (СД2), как и генерализованного метаболического синдрома в целом. Она тесно связана с сердечно-сосудистыми факторами риска, вносящими существенный вклад в развитие ишемической болезни сердца, поэтому для уменьшения риска развития осложнений необходимо не только достижение компенсации углеводного обмена, но и комплексная коррекция остальных метаболических нарушений.
Существует много работ, посвященных эволюции ИР в СД2. Развитие гипергликемии при СД2 связывают как с уменьшением утилизации глюкозы периферическими тканями, так и с повышением продукции глюкозы печенью, т.е. резистентностью печени к действию инсулина, подавляющему образование в ней глюкозы.
Показано влияние на чувствительность к инсулину и генетических особенностей. Так, родственники первой степени родства с нарушенной и даже с нормальной толерантностью к глюкозе имеют выраженную ИР по сравнению с лицами контрольной группы. Несмотря на большое количество исследований, свидетельствующих о наличии генетической предрасположенности к ИР и СД2, природа этих генетических факторов во многом остается невыясненной. Это может быть связано с тем, что развитие заболевания у разных людей обусловлено комбинацией вариантов разных генов, каждый из которых сам по себе имеет небольшой эффект, что затрудняет выявление этих вариантов.
МЕТАБОЛИЗМ ГЛЮКОЗЫ У ЗДОРОВЫХ ЛИЦ И МЕХАНИЗМЫ ЕГО НАРУШЕНИЯ
В норме уровень глюкозы регулируется как инсулинозависимыми, так и инсулиннезависимыми процессами, которые вносят свой вклад как в ее регуляцию натощак, так и в постпрандиальном состоянии [9, 10]. Головной мозг и нервная система являются в основном инсулинонезависимыми; они автономно регулируют потребление глюкозы как энергетического источника с помощью транспортера глюкозы 1 (GLUT-1). Мышечная и жировая ткани являются инсулинозависимыми. В качестве первичного источника энергии они могут использовать как глюкозу, так и кетоновые тела. То, какой вариант энергетического источника будет ими использоваться, первично определяется количеством инсулина, связанного с клеточными инсулиновыми рецепторами. В присутствии большого количества инсулина клетка преимущественно использует глюкозу, активно захватывая и метаболизируя ее или создавая запасы глюкозы в виде гликогена в мышцах или в виде жира в жировой ткани, при этом эффективно снижается уровень постпрандиальной гликемии [11]. Когда уровень инсулина низкий, клетка переключается на метаболизм кетоны/свободные жирные кислоты со снижением утилизации глюкозы, вместо которой в качестве источника энергии используются свободные жирные кислоты, поступающие из кровотока [12]. Желудочно-кишечный тракт (ЖКТ) принимает участие в гомеостазе глюкозы, так как обеспечивает поступление глюкозы в организм при пищеварении. У больных с ИР или НТГ, дополнительное всасывание глюкозы в ЖКТ ухудшает уже нарушенные регуляторные механизмы гомеостаза глюкозы. Кроме того, в ЖКТ в ответ на прием пищи высвобождаются инкретины – гормоны, способствующие снижению постпрандиального уровня гликемии [13]. Инсулин и глюкагон, секретируемые островковым аппаратом поджелудочной железы, регулируют гомеостаз глюкозы. Инсулин секретируется в качестве ответной реакции на повышение уровня глюкозы в плазме крови. Секретированный инсулин подавляет продукцию глюкозы печенью (гликогенолиз и глюконеогенез), стимулирует печеночную утилизацию и хранение глюкозы и регулирует утилизацию глюкозы в мышцах и, в меньшей степени, в жировой ткани [14]. Печень осуществляет две основные функции, которые зависят от уровня инсулина. При низком уровне инсулина, например, при состоянии натощак, печень продуцирует глюкозу при гликогенолизе и глюконеогенезе и высвобождает ее для поддержания нормального уровня гликемии натощак. При умеренном или значительно повышенном уровне инсулина печень прекращает продукцию глюкозы и захватывает глюкозу плазмы с последующим созданием ее запаса в виде гликогена.
В состоянии абсолютного голодания (этот термин употребляется в значении натощак) большая часть глюкозы метаболизируется инсулинонезависимыми тканями: 50% поглощает мозг и 25% утилизируется внутренними органами. Инсулинозависимые ткани, прежде всего мышцы, отвечают за утилизацию оставшихся 25% глюкозы. После поступления глюкозы в кишечник или парентерально этот баланс между УГТ и продукцией глюкозы печенью нарушается. В этом случае поддержание нормального гомеостаза глюкозы зависит от трех очень точно скоординированных процессов: секреция инсулина, УГТ, подавление продукции глюкозы печенью.
Чувствительность периферических тканей к инсулину определяется наличием специфических рецепторов, функция которых опосредует стимулирующее влияние инсулина на УГТ с участием глюкозных транспортеров (GLUT) [15]. Связывание инсулина с рецептором приводит к широкому спектру клеточных реакций. Рецептор выполняет три основные функции: 1) с высокой специфичностью распознает в молекуле места связывания инсулина и осуществляет комплексирование с последним с помощью α-субъединицы; 2) опосредует передачу соответствующего сигнала, направленного на активацию внутриклеточных процессов, путем конформационных изменений и активации тирозинкиназы β-субъединицы; 3) осуществляет эндоцитоз (погружение внутрь клетки) гормонорецепторного комплекса, что приводит к лизосомальному протеолизу инсулина с одновременным возвращением субъединицы к мембране клетки [6].
При СД2 в скелетных мышцах наблюдается нарушение активации инсулинового рецептора. Известно, что нарушение аутофосфорилирования инсулинового рецептора может приводить к прекращению дальнейшего каскада реакций, необходимого для действия инсулина, и ИР скелетных мышц [16]. Однако механизм снижения активности инсулинрецепторной тирозинкиназы при СД2 не ясен. Это связано, скорее, с вторичными метаболическими изменениями, чем с мутацией гена инсулинового рецептора.
Инсулинрецепторная тирозинкиназная активность приводит к аутофосфорилированию инсулинового рецептора и к фосфорилированию других клеточных субстратов. Так называемые белки субстрата инсулинового рецептора, insulin receptor substrates (IRS) играют центральную роль в передаче действия инсулина [17]. Субстраты инсулинового рецептора несут связующую функцию между инсулиновым рецептором и другими внутриклеточными субстратами, такими как, например, фосфоинозитид-3-киназа (PI-3-киназа). При стимуляции инсулином PI-3-киназа превращает фосфоинозитол (PI)-4 или PI-4,5-фосфат в PI-3,4 или PI-3,4,5-фосфат. PI-3,4,5-фосфат с помощью PI-3-киназы обеспечивает адапторный участок для PH-домена серин/треонин специфичной протеинкиназы В (РКВ) и фосфолипид-зависимой киназы (PDK 1 и PDK 2) [18]. РКВ, вероятно, вовлечена в целый ряд тканевых эффектов инсулина, включая стимуляцию поглощения глюкозы, гликолиза, синтеза гликогена и белка. Например, РКВ стимулирует перемещение везикул GLUT-4 к цитоплазматической мембране [19]. При СД2 описано нарушение активации РКВ в скелетных мышцах, несмотря на нормальный уровень этого белка [20]. Другое исследование выявило снижение уровня фосфорилирования IRS-1 и PI-3-киназной активности в скелетных мышцах при СД2 и у худых, и у полных больных, а также у больных с ожирением, но без СД, что, возможно, было связано в 50–60% со снижением экспрессии IRS-1 и р85 PI-3-киназы [21].
После образования вторичного мессенджера активируется транспорт глюкозы. Это происходит с помощью транспортеров глюкозы (GLUT) – белков, расположенных на внутренней поверхности клеточных мембран и обеспечивающих перенос глюкозы внутрь клетки. На сегодняшний день известно 11 членов семейства GLUT, но только 7 из них продемонстрировали транспортную активность [22], с четким определением их на уровне различных органов и тканей.
Как только глюкоза транспортировалась в клетку, инициируется ряд механизмов внутриклеточного метаболизма глюкозы. Глюкоза фосфорилируется глюкокиназой [23] и гексокиназой [24] и затем метаболизируется двумя путями: синтезом гликогена [25] и гликолизом [26]. Происходят эти процессы при участии ферментов, находящихся под контролем инсулина. Наиболее важными являются гликогенсинтаза (контроль образования гликогена) и пируватдегидрогеназа (регуляция окисления глюкозы). Во всех инсулинорезистентных состояниях, включая ожирение и СД2, снижение синтеза гликогена является основным внутриклеточным нарушением, ответственным за дефект действия инсулина. Причем при ожирении с нормальной или нарушенной толерантностью к глюкозе оно может быть частично компенсировано за счет гипергликемии. Дальнейшее прогрессирование НТГ с ожирением в СД2 связано с неспособностью гипергликемии компенсировать этот дефект в инсулинозависимой УГТ. Было также продемонстрировано снижение активности пируватдегидрогеназы в адипоцитах и мышцах больных СД2, хотя многие авторы считают это снижение вторичным по отношению к гипоинсулинемии и повышенному уровню свободных жирных кислот, другие не находят этому доказательств.
Можно предполагать, что у больных с НТГ и началом СД2 имеется слабовыраженная ИР, обусловленная уменьшением числа рецепторов к инсулину. У больных с высокой гипергликемией натощак и выраженной ИР преобладает пострецепторный дефект. Между двумя описанными проявлениями ИР при СД2 относительная значимость рецепторных и пострецепторных нарушений варьирует: по мере усиления ИР нарастает выраженность пострецепторного дефекта.
Метаболический синдром – наиболее частое проявление ИР. Однако понятие состояния ИР гораздо шире. Классическими примерами тяжелой наследуемой ИР являются лепречаунизм, синдром Рабсон-Менденхола, ИР типа А. На чувствительность тканей к инсулину влияют различные факторы: возраст, пол, избыточная масса тела и особенно распределение жировой ткани, артериальное давление, наличие дислипидемии, ишемическая болезнь сердца, а также ряд соматических заболеваний, курение, семейный анамнез по СД, качество питания, низкая физическая активность, злоупотребление алкоголем, психоэмоциональные факторы, лекарственные препараты [27]. ИР встречается не только при СД2, но и при других заболеваниях, сопровождающихся нарушениями обмена веществ. ИР встречается более чем у 25% практически здоровых лиц без ожирения, при этом ее степень выраженности сопоставима с выраженностью ИР, наблюдаемой у больных СД2 [28].
При изучении естественного течения ИР в различных популяциях было установлено, что она представляет собой сочетание двух компонентов: генетического, или наследственного, и приобретенного. В семьях больных СД2 прослеживается ее наследственный компонент. Так, родственники первой степени родства с нарушенной и даже с нормальной толерантностью к глюкозе имеют более выраженную ИР по сравнению с лицами контрольной группы. Аналогичные данные получены при проведении исследований у монозиготных близнецов. Еще одним доказательством генетической предрасположенности к СД2 служит то, что в некоторых этнических группах его распространенность чрезвычайно высока. Например, среди жителей острова Науру (Микронезия) она составляет 40%, а среди индейцев Пима (Аризона, США) превышает 50% [29]. Помимо генов, регулирующих углеводный обмен, влияние на риск развития СД2 могут оказывать и гены, вовлеченные в патогенез ожирения.
Около 80–90% больных СД2 имеют избыточную массу тела или ожирение. Так, при ожирении I степени риск СД2 увеличивается в 2 раза, II степени – в 5 раз, III степени – более чем в 10 раз. Особую роль играет распределение жира [30]. Абдоминальное висцеральное отложение жира связано с нарушением толерантности к глюкозе и ИР, независимо от массы тела [31]. Жировая ткань рассматривается сегодня как один из эндокринных органов, являющихся местом синтеза значительного количества гормонов и биологически активных пептидов, большинство из которых влияют на повышение ИР. Существуют доказательства, что они могут ухудшать передачу инсулинового сигнала и вызывать ИР уже на ранних этапах, на стадии предиабета [32]. В висцеральной ткани повышена секреция гормонов, усиливающих ИР (TNF-α, резистин, висфатин, IL-6 и др.), и одновременно снижена экскреция гормона адипонектина, который снижает ИР [15, 33].
Способность гипергликемии непосредственно нарушать чувствительность к инсулину и секрецию инсулина рассматривается как феномен «глюкозотоксичности» [34]. Хроническая гипергликемия снижает инсулинстимулированную утилизацию глюкозы за счет уменьшения транслокации GLUT-4 в мышечных клетках. Способность свободных жирных кислот ингибировать гликолиз может также способствовать развитию ИР, что определяется термином «липотоксичность» [35]. Свободные жирные кислоты снижают чувствительность к инсулину путем уменьшения транспорта глюкозы и фосфорилирования в мышцах.
МАТЕРИАЛЫ И МЕТОДЫ
Для изучения эволюции ИР нами было обследовано 320 больных с различными нарушениями углеводного обмена, из них 262 пациента с СД2, 36 – с НГН, 22 – с НТГ, находившихся на обследовании и лечении в ФГУ Эндокринологический научный центр. Контрольную группу составили 38 здоровых лиц. Исходно 136 пациентов были на диетотерапии, 147 пациентов получали различные пероральные сахароснижающие препараты (ПCСП), 37 пациентов находились на инсулинотерапии. Среди обследованных было 142 мужчины и 216 женщин, возраст больных составил в среднем 52,8±11,0 лет (21–77 лет). Продолжительность заболевания была в среднем 6,7±6,8 лет (0–25 лет), у 128 пациентов нарушение углеводного обмена было выявлено впервые. Средний ИМТ у больных составил 29,8±4,9 кг/м2 (18,4–45,9 кг/м2). Нормальную массу тела (ИМТ<25 кг/м2) имели 16,4% больных, избыточный вес (ИМТ 25–30 кг/м2) – 39,4%, ожирение (ИМТ>30 кг/м2) – 44,2% пациентов. Большинство пациентов имели распределение жировой ткани по абдоминальному типу.
Определение уровня общего холестерина, триглицеридов, холестерина липопротеидов высокой плотности (ЛПВП), холестерина липопротеидов низкой плотности (ЛПНП) проводили на биохимическом анализаторе «Spectrum» фирмы «Abbott» (США) стандартными наборами фирмы, гликированного гемоглобина (HbA1c) и микроальбуминурии – на анализаторе DCA2000+ фирмы «Bayer» (Германия), гликемии – на анализаторе «Reflotron» фирмы «Boehringer Mannheim» (Германия).
Для определения содержания С-пептида и иммунореактивного инсулина (ИРИ) в сыворотке крови использовался электрохемилюминесцентный иммуноанализ (ECLIA) с использованием наборов реактивов «Elecsys C-peptide» и «Elecsys Insulin» фирмы «Roche Diagnostics» (Швейцария). Иммуноферментным методом, используя коммерческие наборы, натощак в сыворотке крови определяли уровень проинсулина (Proinsulin ELISA, «Mercodia»). Иммуноферментным методом, используя коммерческие наборы, натощак в сыворотке крови определяли уровень лептина (Leptin ELISA, «DBC», Канада), адипонектина (Human Adiponectin ELISA, «BioVendor», Чехия), резистина (Human Resistin ELISA, «BioVendor», Чехия), грелина (Total Ghrelin ELISA, «DSL ACTIVE», США), висфатина (Human Visfatin ELISA, «BioSource», США), фактора некроза опухолей-альфа (Human TNF-α, «Bender MedSystems», Австрия). Содержание С-реактивного белка натощак в сыворотке крови определялось иммунотурбидиметрическим методом с использованием набора «CRP (Latex) HS COBAS» фирмы «Roche Diagnostics» (Швейцария).
Определение чувствительности периферических тканей к инсулину проводилось с использованием гиперинсулинемического эугликемического клэмп-метода [36]. Метод основан на непрерывном внутривенном введении инсулина и глюкозы (рис. 1). Скорость инфузии инсулина являлась постоянной и составляла 1 мЕд/кг/мин, точность скорости введения инсулина обеспечивалась шприцевым дозатором «Pilot A2» фирмы «Fresеnius Vial» (Франция-Германия). Скорость введения глюкозы изменялась таким образом, чтобы поддерживать целевой уровень гликемии (5,3±0,3 ммоль/л), точность скорости введения глюкозы обеспечивалась волюметрическим дозатором «INCA-ST» фирмы «Fresеnius Vial» (Франция-Германия). Общая продолжительность исследования составляла 4–6 ч. Скорость введения глюкозы в равновесном состоянии определяла скорость УГТ, что использовалось для вычисления коэффициента утилизации (М-индекс), как среднего арифметического из 10–12 дискретных значений скорости инфузии глюкозы, деленное на массу тела обследуемого или на нежировую массу тела, за 1 минуту.
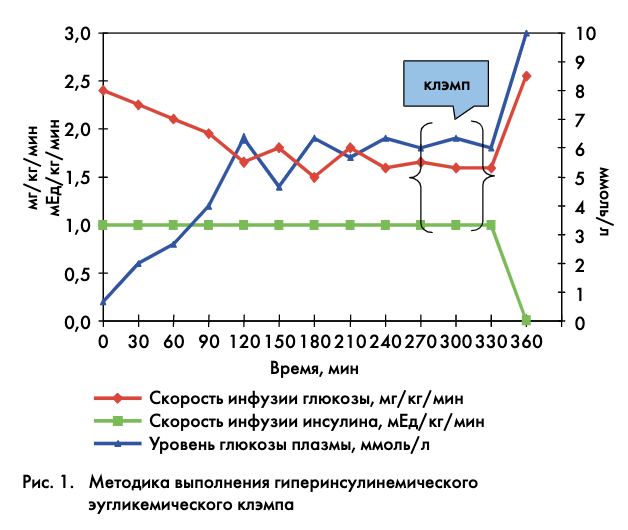
Также оценка уровня ИР проводилась с помощью структурной математической модели на основе определения инсулина и глюкозы плазмы натощак (ГПН) – HOMA (homeostasis model assesment) – с вычислением коэффициентов ИР и секреции инсулина [37, 38].
- Индекс ИР (HOMA-ИР)= ИРИ (мкЕд/мл) х ГПН (ммоль/л) / 22,5
- Функциональная активность бета-клеток (HOMA-ФБ)= 20 х ИРИ (мкЕд/мл) / ГПН (ммоль/л) - 3,5
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Связь чувствительности к инсулину с основными клиническими и биохимическими показателями у больных с различными нарушениями углеводного обмена
При исследовании степени нарушения чувствительности к инсулину у всех лиц с нарушениями углеводного обмена значение М-индекса при исходном обследовании варьировало от 0,15 до 12 мг/кг/мин, составляя в среднем 4,01±2,27 мг/кг/мин, что почти в 2 раза ниже показателей здоровых лиц (7,72±1,89 мг/кг/мин, р<0,001). Анализ распределения частот встречаемости показал, что у 20,4% пациентов М-индекс находился в пределах 2 мг/кг/мин, что можно расценивать как выраженное снижение чувствительности к инсулину. У 31,9% М-индекс был в интервале от 2 до 4 мг/кг/мин, что соответствует умеренному снижению, у 27,2% – в интервале от 4 до 6 мг/кг/мин – незначительное снижение, у 20,5% периферическая чувствительность соответствовала близким к нормальным показателям (выше 6 мг/кг/мин). Значения М-индекса были одинаковыми у мужчин (3,96±2,20 мг/кг/мин) и женщин (4,13±2,29 мг/кг/мин). Это противоречит некоторым литературным данным о влиянии пола на чувствительность к инсулину у здоровых лиц и в общей популяции больных СД2 [39]. Поскольку в литературе есть указания на возможность возрастных изменений чувствительности к инсулину [40], мы проанализировали значения скорости УГТ у больных с нарушениями углеводного обмена различных возрастных групп. Было показано, что у молодых больных (до 30 лет), особенно у мужчин, скорость УГТ несколько выше, но в связи с небольшим числом этих пациентов различие индекса M не было статистически достоверным. Уровень М-индекса не зависел от длительности (r=0,13, р=0,087) и возраста дебюта заболевания (r=0,06, р>0,3).
При сопоставлении уровня ИР и массы тела больных была обнаружена достоверная взаимосвязь между ИМТ и М-индексом в общей группе (r=-0,31, р<0,0001). Большинство исследований подтверждают, что и общая жировая масса, и распределение по абдоминальному типу ассоциированы с ИР [41]. Это подтверждается и улучшением чувствительности к инсулину в нашем исследовании при повторном ее исследовании у части больных с впервые выявленным СД после снижения массы тела: на фоне уменьшения ИМТ с 29,4±0,5 до 28,0±0,5 кг/м2 (p<0,01) скорость УГТ возросла с 2,11±0,42 до 4,73±1,13 мг/кг/мин (p<0,001). Также показана выраженная обратная зависимость М-индекса от окружности талии (ОТ) (r=-0,35, р=0,003) и меньшая – от окружности бедер (ОБ) (r=-0,23, р=0,047) и их соотношения (ОТ/ОБ) (r=-0,23, р=0,049), что отражает влияние прежде всего степени висцерального ожирения на уровень чувствительности к инсулину.
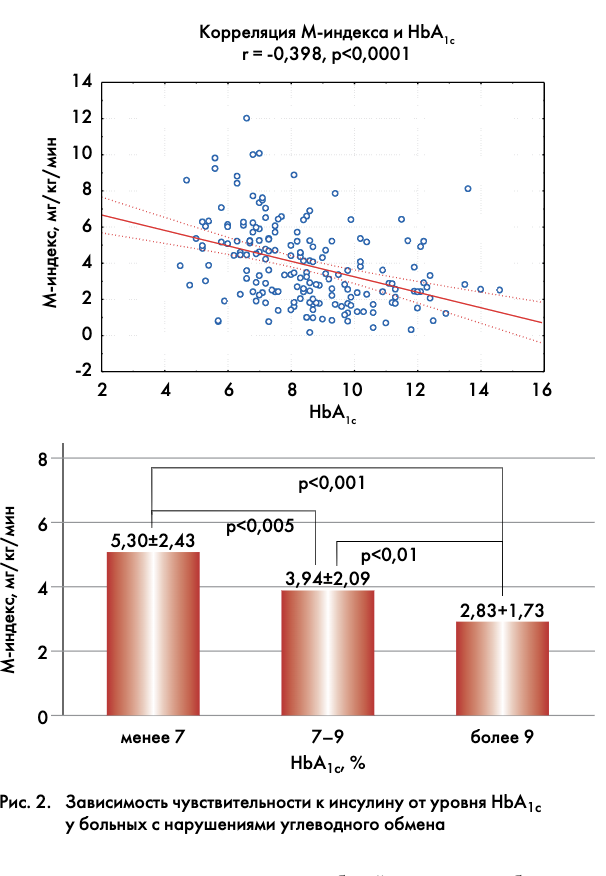
Была выявлена сильная обратная корреляции скорости УГТ от степени компенсации углеводного обмена: уровня HbA1c (r=-0,40, р<0,0001) и уровня ГПН (r=-0,43, р<0,0001) (рис. 2). Пациенты, достигшие целевого уровня HbA1c (менее 7%), имели достоверно более высокий М-индекс по сравнению с больными, имеющими выраженную декомпенсацию (HbA1c выше 9%): 5,30±2,43 и 2,83±1,73 мг/кг/мин, соответственно (р<0,001). Таким образом, можно сказать, что глюкозотоксичность играет существенную роль в развитии и усугублении ИР, что подтверждается данными литературы, в том числе о влиянии снижения гликемии на улучшение чувствительности к инсулину [34, 42]. В нашем исследовании степень снижения HbA1c была взаимосвязана с чувствительностью к инсулину при повторном определении – наибольшее снижение HbA1c, например, на фоне инсулинотерапии, сопровождалось наименьшей ИР (r=-0,59, р<0,01).
Было продемонстрировано, что существует достоверная обратная корреляция между уровнем триглицеридов и М-индексом (r=-0,31, р<0,0001). Пациенты, достигшие целевого уровня триглицеридов (менее 1,7 ммоль/л), рекомендованного отечественными стандартами лечения СД [43], имели достоверно более высокий М-индекс по сравнению с больными, имеющими выраженную гиперлипидемию (уровень триглицеридов выше 2,2 ммоль/л): 4,30±2,45 и 2,85±1,62 мг/кг/мин, соответственно (р<0,001). Корреляция с уровнем холестерина была достоверной, но значительно слабее (r=-0,18, р=0,018). Не было отмечено достоверной корреляции с уровнями ЛПНП (r=-0,05, р=0,511) и ЛПВП (r=0,19, р=0,074).
В таблице 1 показана сравнительная характеристика больных с различными нарушениями углеводного обмена и здоровых лиц. Было отмечено не только достоверное отличие скорости УГТ во всех группах больных по сравнению с контрольной группой, но и отличие пациентов с СД2 по сравнению с лицами с НТГ И НГН. Среди пациентов с нарушениями углеводного обмена лица с НТГ имели самый молодой возраст (41,5±11,4 года) и, соответственно, самое раннее начало заболевания (40,1±11,4 года). Они не отличались между собой по ИМТ, ОБ, ОТ, уровню общего холестерина, ЛПВП и ЛПНП холестерина, триглицеридов. Хотя диагноз НТГ включает в себя как нормальный уровень ГПН, так и соответствующий НГН, у обследованных лиц с НТГ уровень ГПН был достоверно ниже по сравнению с лицами с НГН (5,38±1,25 vs 6,54±0,41 ммоль/л, р=0,018). Было отмечено не только достоверное отличие скорости УГТ во всех группах больных по сравнению с контрольной группой, но и отличие пациентов с СД2 по сравнению с лицами с НТГ и НГН. Больные с СД2 имели почти в 2 раза меньший М-индекс по сравнению со здоровыми лицами: 3,75±2,20 vs 7,72±1,89 мг/кг/мин (р<0,0001). Пациенты с ранними нарушениями углеводного обмена продемонстрировали М-индекс в 1,3–1,4 раза меньше по сравнению с группой контроля. При анализе частоты встречаемости показателей скорости УГТ у пациентов с различными нарушениями углеводного обмена выявлено следующее: в отличие от группы пациентов с СД2, в группах с НТГ и НГН не встречалось значений М-индекса ниже 2 мг/кг/мин. Нормальные показатели были только у 17% больных СД2 и у 46% и 43% больных с НТГ и НГН, соответственно. В группе с НТГ почти в 2 раза чаще отмечалось умеренное снижение чувствительности к инсулину (М-индекс от 2 до 4 мг/кг/мин), чем в группе НГН: 31 и 14%, соответственно.
При анализе взаимосвязи периферической чувствительности к инсулину у больных СД2 не было отмечено достоверной корреляции скорости УГТ с возрастом дебюта СД (r=0,07, р>0,3), ОБ (r=-0,19, р>0,1), ОТ/ОБ (r=-0,24, p=0,055), уровнем холестерина (r=-0,15, р=0,069). Так же как и в целом у всех больных с различными нарушениями углеводного обмена отмечена сильная обратная корреляция М-индекса с ИМТ (r=-0,29, p<0,001), ОТ (r=-0,33, р=0,008), HbA1c (r=-0,39, p<0,0001), ГПН (r=-0,34, p<0,0001), уровнем триглицеридов (r=-0,37, p<0,0001). Несколько парадоксальной выглядит умеренная прямая корреляция скорости УГТ с возрастом (r=0,22, p=0,005) и длительностью заболевания (r=0,24, p=0,002), что, скорее всего, связано с применением у части больных препаратов, влияющих на чувствительность к инсулину. Подтверждением этого служит тот факт, что у больных с впервые выявленным СД2 такой корреляции обнаружено не было.
Для изучения роли снижения чувствительности к инсулину в развитии СД были проанализированы различия в клинических и биохимических показателях у больных СД2, которые были разделены на 5 групп в зависимости от степени ИР. В 1-ю группу вошли больные с выраженным снижением чувствительности к инсулину (коэффициент М менее 2 мг/кг/мин), во 2-ю – имеющие М в интервале от 2 до 4 мг/кг/мин (умеренное снижение), в 3-ю – больные с М от 4 до 6 мг/кг/мин (незначительное снижение), в 4-ю – больные с М от 6 до 8 мг/кг/мин (нормальная чувствительность), в 5-ю – пациенты с высокой чувствительностью (М-индекс выше 8 мг/кг/мин). Результаты этого анализа представлены в таблице 2. Больные с более низким М-индексом характеризовались более высокими ИМТ, ОТ и ОТ/ОБ, что еще раз отражает влияние висцерального ожирения на развитие ИР. Глюкозотоксичность негативно влияет на чувствительность периферических тканей к инсулину: уровень HbA1c и ГПН были выше в группе больных СД2 с низкими значениями скорости УГТ. Также была подтверждена и липотоксичность, выражающаяся в более высоком уровне триглицеридов у пациентов с низкими значениями М-индекса.
УРОВНИ ГОРМОНОВ И ЦИТОКИНОВ И ИХ СВЯЗЬ С ПОКАЗАТЕЛЯМИ ЧУВСТВИТЕЛЬНОСТИ К ИНСУЛИНУ
При исследовании ряда гормонов и цитокинов у больных с различными нарушениями углеводного обмена было выявлено, что уровни адипонектина и резистина у больных СД2 были ниже по сравнению со здоровыми лицами, а уровни фактора некроза опухолей-альфа (TNF-α) и проинсулина– выше (таблица 3). Как известно из литературных источников, уровень адипонектина положительно коррелирует с чувствительностью к инсулину и отрицательно – с массой висцеральной жировой ткани [6]. В нашем исследовании была отмечена достоверная прямая корреляция уровня адипонектина с М-индексом (r=0,24, p=0,019) и слабая обратная корреляция – с ОТ (r=-0,18, p=0,036) ОТ/ОБ (r=0,21, p=0,041). Достоверной корреляции с ИМТ выявлено не было. Механизм влияния на чувствительность периферических тканей к инсулину заключается прежде всего в том, что адипонектин снижает поступление НЭЖК в печень и стимулирует их окисление путем активации протеинкиназы, способствуя сокращению продукции глюкозы печенью. Уровни лептина и висфатина достоверно не различались. Известно, что экспрессия лептина напрямую зависит от содержания липидов в клетках, в большей степени в подкожной жировой ткани. Хотя секреция лептина жировой тканью сопровождается повышением ИР [44], мы не обнаружили достоверной корреляция уровня лептина и М-индекса. Подобные различия для адипонектина, резистина и TNF-α отмечены и на стадии ранних нарушений углеводного обмена. Но следует отметить, что достоверных различий в уровне данных гормонов между больными с различными нарушениями углеводного обмена не было. Только уровень висфатина в группе лиц с НТГ был ниже по сравнению с больными СД2 и НГН. Также продемонстрирован достоверно более высокий уровень высокоспецифичного С-реактивного белка (СРБ) как у больных СД2, так и у лиц с НГН и НТГ по сравнению с контрольной группой.
ВЛИЯНИЕ РАЗЛИЧНЫХ ПРЕПАРАТОВ НА ЧУВСТВИТЕЛЬНОСТЬ К ИНСУЛИНУ
Среди препаратов, влияющих на чувствительность к инсулину, на первом этапе лечения СД2 рассматривается метформин. В нашей работе метформин был назначен 16 пациентов с впервые выявленным СД2, НТГ и НГН в возрасте 34–56 лет (средний возраст 47,5±9,0 лет) с ИМТ от 22,6 до 45,9 кг/м2 (средний 31,6±6,7 кг/м2). Все пациенты не получали сахароснижающей терапии, гликемический контроль не соответствовал целевым параметрам. Исследование проводилось до и через 3 мес после лечения метформином в дозе 850–1700 мг. Средний уровень HbA1c снизился с 7,6±2,1 до 6,2±0,9% (р<0,01); 50% больных достигли целевых значений HbA1c (до 6,5%). Хорошо известно, что бигуаниды оказывают существенное влияние не только на нормализацию углеводного обмена, но также улучшают состояние липидного обмена [45, 46]. В изучаемой группе пациентов уровень холестерина снизился с 5,61±1,09 до 5,07±1,19 ммоль/л (р<0,05), 58% больных достигли нормы. Уровень триглицеридов снизился с 2,36±1,8 до 1,6±1,0 ммоль/л (р<0,02), 83% достигли нормы. М-индекс, характеризующий степень ИР, у больных СД2 увеличился с 4,10±1,60 до 5,82±2,18 (р<0,01); в группе пациентов с НТГ и НГН – с 3,68±0,91 до 6,98±2,24 (р<0,001). Уровень СРБ снизился с 3,55±3,42 до 2,35±2,40 мг/л (р<0,01), висфатина – с 3,35±2,56 до 2,46±1,50 нг/мл (р<0,02). Достоверного изменения уровней адипонектина, лептина, резистина, грелина не отмечалось. Таким образом, полученные данные подтверждают возможность использования метформина до развития СД2, на стадиях нарушенной регуляции глюкозы, что было ранее показано в отношении профилактики СД2 [47].
Препараты тиазолидиндионового ряда нашли свое применение последнее десятилетие. Соединения этого класса выступают в роли агонистов ядерных гамма-рецепторов, активируемых пролифераторами пероксисом (PPAR-γ). Активация PPAR-γ рецепторов модулирует транскрипцию ряда генов, связанных с передачей эффектов инсулина на клетки и участвующих в контроле уровня глюкозы и метаболизме липидов [48]. Нами было проведено открытое, проспективное наблюдательное исследование эффективности и безопасности применения препарата пиоглитазон у пациентов с СД2. В исследование был включен 81 пациент: в группу монотерапии – 28, в группу комбинированной терапии – 53 пациента. В группу монотерапии включали пациентов, которые до этого находились на диетотерапии и не получали пероральных сахароснижающих препаратов (ПССП). В группу комбинированной терапии включали пациентов, получавших один из ПССП (глибенкламид или метформин), но не имевших удовлетворительного гликемического контроля. Пиоглитазон назначался всем пациентам в дозе 30 мг 1 р/сут, продолжительность периода активной терапии в обеих группах составила 3 мес. После терапии пиоглитазоном было зарегистрировано улучшение показателей гликемического контроля во всех группах терапии. В группе монотерапии статистически достоверно снизились уровень HbA1c на 1,3±1,2% (с 8,6±1,4 до 7,3±1,2%), уровень ГПН на 1,6±2,2 ммоль/л (с 10,2±2,8 до 8,6±2,2 ммоль/л) и показатель HOMA-ИР на 3,2±5,4 (с 10,6±6,4 до 7,4±3,8). Кроме того, наблюдалось увеличение HOMA-ФБ на 9,7±60,4 и снижение уровня ИРИ на 4,1±12,2 мкЕд/мл, однако данные изменения не были статистически значимыми. В группе комбинированной терапии статистически достоверно снизился уровень HbA1c на 0,8±0,8% (с 8,4±1,2 до 7,6±1,1%), уровень ГПН на 1,7±2,3 ммоль/л (с 9,9±2,7 до 8,2±2,0 ммоль/л), значение HOMA-ИР на 3,7±5,7 (с 9,3±5,9±6,4 до 5,6±2,9) и уровень ИРИ на 5,5±9,9 мкЕд/мл (с 20,8±11,4 до 15,3±6,4 мкЕд/мл). Отмечалось увеличение HOMA-ФБ и в данной группе пациентов на 3,0±44,6, но оно также не было статистически достоверным. Анализ подгрупп комбинированной терапии показал, что в обеих подгруппах наблюдалось статистически достоверное снижение уровня HbA1c, ГПН, HOMA-ИР и ИРИ, но не отмечалось статистических различий между подгруппами по всем анализируемым показателям в конце терапии.
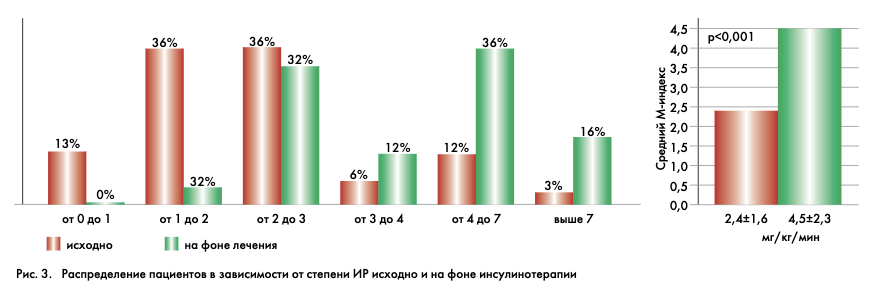
Для оценки влияния инсулинотерапии на показатели чувствительности к инсулину было обследовано 43 пациента с СД2 до и после назначения инсулина. Возраст больных составил в среднем 56,1±8,6 лет (38–75 лет), длительность заболевания была в среднем 11,7±6,8 лет (4–31 год). Средний ИМТ у больных составил 29,5±5,3 кг/м2 (20,2–42,1 кг/м2). Для сравнения влияния моно- и комбинированной инсулинотерапии больные были рандомизированы на 3 группы: монотерапия инсулином в интенсифицированном режиме (n=20), комбинация с приемом метформина в дозе 1500 мг/сут (n=11) и пиоглитазоном 30 мг/сут (n=12). Повторное обследование осуществляли через 6 мес после перевода на инсулинотерапию. Значение М-индекса при исходном обследовании составило в среднем 2,4±1,6 мг/кг/мин, что в 3 раза ниже показателей здоровых лиц.
После 6 мес инсулинотерапии, при повторном определении скорости УГТ, отмечено ее достоверное увеличение в 2 раза – до 4,5±2,3 мг/кг/мин (р<0,001). Значительно изменилось распределение частоты встречаемости отдельных значений (рис. 3). На фоне инсулинотерапии не выявлено показателей ниже 1 мг/кг/мин, в 2 раза (до 36%) уменьшилось количество пациентов, имеющих выраженное снижение М-индекса (от 1 до 3 мг/кг/мин), 48% имели чувствительность к инсулину от 3 до 7 мг/кг/мин (vs 25% исходно), в 5 раз (до 16%) увеличилась доля лиц с нормальной скоростью УГТ.
Увеличение массы тела на фоне инсулинотерапии составило в среднем 2,7 кг, ИМТ увеличился до 30,3±4,2 кг/м2 (р<0,05), соотношение ОТ/ОБ в целом по группе не изменилось – 0,9±0,1. Степень увеличения массы тела зависела от уровня ИР и была максимальной при исходном М-индексе менее 1 и более 4 мг/кг/мин, по сравнению с группами 1–2 и 2–4 мг/кг/мин. Степень изменения скорости УГТ определялась исходным уровнем периферической чувствительности к инсулину (r=-0,55, р<0,01). В группе с исходным М-индексом менее 1 мг/кг/мин наблюдалось наибольшее увеличение среднего показателя (с 0,4±0,3 до 2,8±2,0, р<0,05), в то время как у пациентов с М-индексом более 4 мг/кг/мин достоверного изменения не отмечено (с 6,1±1,7 до 4,6±1,6, р>0,05), несмотря на одинаково выраженное увеличение массы тела в обеих группах. Продемонстрирована обратная зависимость степени увеличения массы тела с исходным ИМТ (r=-0,39, р<0,05). Наибольшие значения М-индекса на фоне инсулинотерапии наблюдались у больных с исходно нормальной массой тела: 7,2±2,3 vs 3,5±1,6; 4,1±1,9 мг/кг/мин, р<0,01. В процессе лечения средний уровень ГПН снизился с 13,3±3,1 до 9,6±2,4 ммоль/л (р<0,01), средний уровень HbA1c – с 11,2±1,6 до 7,7±1,4% (р<0,01). Наибольшее снижение HbA1c сопровождалось наименьшей ИР на фоне инсулинотерапии (r=-0,59, р<0,01), что отражает непосредственное влияние гипергликемии (глюкозотоксичность) на чувствительность к инсулину. Назначение инсулина приводило к значительному улучшению липидного профиля: снижению общего холестерина (5,4±1,1 ммоль/л, р<0,001), ЛПНП (3,4±1,0 ммоль/л, р=0,001) и триглицеридов (1,62±0,7 ммоль/л, р<0,001) Уровень ЛПВП холестерина достоверно не изменился. Корреляция наблюдалась только с уровнем триглицеридов (r=-0,53, p<0,01). Показательно то, что изменения зафиксированы, несмотря на увеличение массы тела обследуемых за время инсулинотерапии. В группе больных с наименьшей степенью ИР показатели липидного профиля достигли значений, соответствующих низкому риску развития сосудистых осложнений.
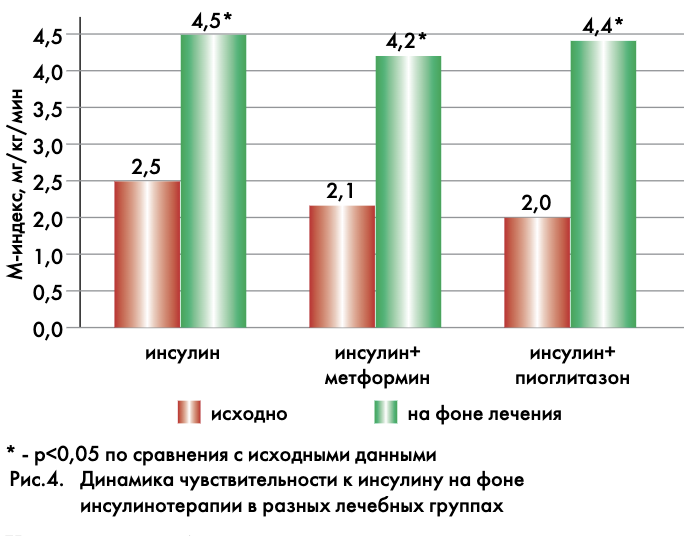
Во всех 3 группах (моноинсулинотерапия, комбинация с метформином 1500 мг/сут и пиоглитазоном 30 мг/сут) инсулинотерапия проводилась в интенсифицированном режиме, что было обусловлено степенью ухудшения метаболического контроля. При сравнении групп до начала инсулинотерапии не выявлено различий по ИМТ, показателям С-пептида, а также уровню липидов, гликированного гемоглобина и чувствительности к инсулину. На фоне инсулинотерапии во всех группах отмечена положительная динамика показателей метаболического контроля, достоверных отличий между группами при этом не наблюдалось. Улучшение чувствительности к инсулину на фоне инсулинотерапии произошло во всех лечебных группах в равной степени: в 1-й группе – в 1,8 раз, во 2-й – в 2 раза и в 3-й – в 2,2 раза, р>0,1 между группами (рис. 4). Среднесуточные дозы инсулина у больных различных групп достоверно не отличались
(0,66±0,2 vs 0,59±0,1 vs 0,57±0,6 Ед/кг/сут).
Таким образом, было показано наличие ИР у больных с впервые выявленным СД2, НТГ и НГН, которое проявляется в снижении скорости утилизации глюкозы тканями, измеренной клэмп-методом. При этом отмечалось более выраженное снижение чувствительности к инсулину при СД2, чем при НТГ и НГН (в среднем на 50, 25 и 15% соответственно по сравнению со здоровыми лицами). Изменение уровня ИР в процессе эволюции СД2 носит вторичный характер, связанный с глюкозотоксичностью, липотоксичностью, увеличением массы тела, проводимой сахароснижающей терапией. Все это диктует необходимость интенсифицировать сахароснижающую терапию, в том числе переводить на инсулинотерапию, не дожидаясь выраженного ухудшения чувствительности к инсулину. При изучении влияния гормонов и цитокинов на показатели ИР выявлено достоверное отличие в уровнях адипонектина, резистина, проинсулина и TNF-α между здоровыми лицами и пациентами с различными нарушениями углеводного обмена. Назначение инсулинотерапии приводит к двукратному увеличению чувствительности к инсулину у исследуемой категории больных в прямой зависимости от степени улучшения гликемического контроля и при сопутствующем снижении атерогенности липидного профиля.
СПИСОК ЛИТЕРАТУРЫ
- Himsworth H.P., Kerr R.B. Insulin-sensitive and Insulin-insensitive types diabetes mellitus // Clin. Sci. - 1939. - № 4. - Р. 119-152.
- Дедов И.И., Шестакова М.В. Сахарный диабет. - М.: Универсум Паблишинг. - 2003.
- Del Prato S., Leonetti F., Matsuda M., DeFronzo R.A. et al. Effect of sustained physiologic hyperinsulinemia and hyperglycaemia on insulin secretion and insulin sensitivity in man // Diabetologia. - 1994. - № 37. - Р. 1025-1035.
- Балаболкин М.И., Клебанова Е.М. Инсулинорезистентность в патогенезе сахарного диабета 2 типа // Сахарный диабет. - 2001. - № 1. - С. 28-37.
- Шестакова М.В., Брескина О.Ю. Инсулинорезистентность: патофизиология, клинические проявления, подходы к лечению // Consilium medicum. - 2002. - № 10.
- Kumar S., O'Rahily S. Insulin Resistance. Insulin action and its disturbances in diseases // John Wiley & Sons, Ltd. - 2005.
- Nathan D.M., Davidson M.B., DeFronzo R.A. et al. Impaired fasting glucose and impaired glucose tolerance // Diabetes Care. - 2007. - № 30. - Р. 753-759.
- Reaven G.M. Role of insulin resistance in human disease (syndrome X): an expanded definition // Annual Review of Medicine. - 1993. - № 44. - Р. 121-131.
- Балаболкин М.И. Диабетология. - М.: Медицина. - 2000.
- DeFronzo R.A. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM // Diabetes. - 1988. - № 37. - Р. 667-687.
- Abel E.D., Peroni O., Kim J.K., Kim Y.В., Boss O., Hadro E., Minnemann Т., Shulman G. I., Kahn В. B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver // Nature. - 2001. - № 409. - Р. 729-733.
- Roden M., Price Т.В., Perseghin G., Petersen K.F., Rothman D.L., Cline G.W., Shulman G.I. Mechanism of free fatty acid-induced insulin resistance in humans // J. Clin. Invest. - 1996. - № 97. - Р. 2859-2865.
- Elrick H., Stimmler L., Hlad C.J., Turner D.A. Plasma insulin responses to oral and intravenous glucose administration // J. Clin. Endocrinol Metab. - 1964. - № 24. - Р. 1076-1082.
- Clark M.G., Wallis M.G., Barrett E.J., Vincent M.A., Richards S.M., Clerk L.H., Rattigan S. Blood flow and muscle metabolism: a focus on insulin action // Am. J. Physiol. - 2003. - № 284. - Р. E241-258.
- Дедов И.И., Балаболкин М.И. Инсулиновая резистентность в патогенезе сахарного диабета типа 2 и медикаментозная возможность ее преодоления // Врач. - 2006. - № 11.
- Olefsky J.M. The insulin receptor. A multifunctional protein // Diabetes. - 1990. - № 39. - Р. 1009-1016.
- White M.F. IRS proteins and the common path to diabetes // Am J Physiol Endocrinol Metab. - 2002. - № 283. - Р. E413-422.
- Kimber W.A., Deak M., Prescott A.R., Alessi D.R. Interaction of the protein tyrosine phosphatase PTPL1 with the PtdIns(3,4)P2-binding adaptor protein TAPP1 // Biochem. J. - 2003. - № 376. - Р. 525-535.
- Heller-Harrison R.A., Morin M., Guilherme A., Czech M.P. Insulinmediated targeting of phosphatidylinositol 3-kinase to GLUT4-containing vesicles // J. Biol. Chem. - 1996. - № 271. - Р. 10200-10204.
- Whiteman E.L., Cho H., Birnbaum M.J. Role of Akt/protein kinase В in metabolism // Trends Endocrinol Metab. - 2002. - № 13. - Р. 444-451.
- Ueki K., Fruman D.A., Yballe С.M., Fassaur M., Klein J., Asano Т., Cantley L.C., Kahn C.R. Positive and negative roles of p85alpha and p85beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling // J. Biol. Chem. - 2003. - № 278. - Р. 48453-48466.
- Joost H. G., Bell G. I., Best J.D., Birnbaum M.J., Charron M.J., Chen Y.Т., Doe ge H., James D.E., Lodish H.F., Moley К.H. et al. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators // Am. J. Physiol. - 2002. - № 282. - Р. E974-976.
- Barzilai N., Rossetti L. Role of glucokinase and glucose-6-phosphatase in the acute and chronic regulation of hepatic glucose fluxes by insulin // Biol. Chem. - 1993. - № 268. - Р. 25019-25025.
- Printz R.L., Koch S., Potter L.R., O'Doherty R.M., Tiesinga J. J., Moritz S., Granner D.K. Hexokinase II mRNA and gene structure, regulation by insulin, and evolution // J. Biol. Chem. - 1993. - № 268. - Р. 5209-5219.
- Cohen P., Frame S. The renaissance of GSK3 // Nat. Rev. Mol. Cell. Biol. - 2001. - № 2. - Р. 769-776.
- Harris R.A., Bowker-Kinley M.M., Huang B., Wu P. Regulation of the activity of the pyruvate dehydrogenase complex // Adv. Enzyme Regul. - 2002. - № 42. - Р. 249-259.
- Kahn S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the patophysiology of Type 2 diabetes // Diabetologia. - 2005. - № 48. - Р. 3-19.
- Reaven G.M. Role of insulin resistance in human disease // Diabetes. - 1988. - № 37. - Р. 1595-1607.
- Cassell P.G., Jackson A.E., North B.V., Evans J.C., Syndercombe-Court D., Phillips C., Ramachandran A., Snehalatha C., Gelding S.V., Vijayaravaghan S., Curtis D., Hitman G.A. Haplotype combinations of calpain 10 gene polymorphisms associate with increased risk of impaired glucose tolerance and type 2 diabetes in South Indians // Diabetes. - 2002. - № 51. - Р. 1622-1628.
- Haffner S.M., Karhapaa P., Mykkanen L., Laakso M. Insulin resistance, body fat distribution, and sex hormones in men // Diabetes. - 1994. - № 43. - Р. 212-219.
- Peiris A.N., Struve M.F., Mueller R.A., Lee M.B., Kissebah A.H. et al. Glucose metabolism in obesity: influence of body fat distribution // J. Clin. Endocrinol. Metab. - 1988. - № 67 (4). - Р. 760-767.
- Jazet I. M., Pijl H., Meinders A. E. Adipose tissue as an endocrine organ: impact on insulin resistance // Neth. J. Med. - 2003. - № 61 (6). - Р. 194-212.
- Fasshauer M., Paschke1 R. Regulation of adipocytokines and insulin resistance // Diabetologia. - 2003. - № 46. - Р. 1594-1603.
- Yki-Järvinen H. Glucose toxicity // Endocr. Rev. - 1992. - № 11. - Р. 415-431.
- McGarry J.D., Dobbins R.L. Fatty acids, lipotoxicity and insulin resistance // Diabetologia. - 1999. - № 42. - Р. 128-138.
- DeFronzo R.A., Tobin J.D., Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance // American Journal of Physiology. - 1979. - № 237 (3). - Р. E214-223.
- Matthews D.R., Hosker J.P., Rudenski A.S., Turner R.C. et al. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentration in man // Diabetologia. - 1985. - № 28. - Р. 412-419.
- Wallace T.M., Levy J.C., Matthews D.R. Use and abuse of HOMA modeling // Diabetes Care. - 2004. - № 27. - Р. 1487-1495.
- Nuutila P., Knuuti M.J., Maki M., Yki-Jarvinen H. et al. Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography // Diabetes. - 1996. - № 44 (1). - Р. 31-36.
- DeFronzo R.A., Bonadonna R.S., Ferrannini E. Pathogenesis of NIDDM. A balanced overview // Diabetes Care. - 1992. - № 15. - Р. 318- 368.
- Frayn K.N. Visceral fat and insulin resistance - causative or correlative? // Br J Nutr. - 2000. - № 83 (Suppl. 1). - S71-77.
- Rossetti L. Glucose toxicity: the implications of hyperglycemia in the pathophysiology of diabetes mellitus // Clin. Invest. Med. - 1995. - № 18. - Р. 255-260.
- Алгоритмы специализированной медицинской помощи больным сахарным диабетом (под редакцией Дедова И.И., Шестаковой М.В.). Издание четвертое, дополненное. - М., 2009. - 103 с.
- Pittas A.G., Joseph N.A. Adipocytokines and insulin resistance // J. Clin. Endocrinol. Metab. - 2004. - № 89. - Р. 447-452.
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) // Lancet. - 1998. - № 352. - Р. 854-865.
- United Kingdom Prospective Diabetes Study 24: a 6-years, randomized, controlled trial comparing sulfonylurea, insulin and metformin therapy in patients with newly diagnosed type 2 diabetes that could not be controlled with diet therapy // Ann. Intern. Med. - 1998. - № 128 (3). - Р. 165-175.
- Knowler W.C., Barrett-Connor E., Fowler S.E. et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin // N.Engl. J. Med. - 2002. - № 346. - Р. 393-403.
- Semple R.K., Chatterjee V.K., O'Rahilly S. PPAR gamma and human metabolic disease // J. Clin. Invest. - 2006. - № 116. - Р. 581-589.
Похожие статьи
Квиткова Л.В.
Халимова А.С.
Юлия Эдуардовна Азарова
Елена Юрьевна Клёсова
Александр Иванович Конопля
Юлия Эдуардовна Азарова
Елена Юрьевна Клёсова
Светлана Юрьевна Сакали
Алексей Павлович Ковалев
и другие.
Иван Иванович Дедов
Марина Владимировна Шестакова
Александр Сергеевич Аметов
Михаил Борисович Анциферов
и другие.
Всеволод Арсеньевич Ткачук
Александр Вячеславович Воротников