Эта статья опубликована под лицензией Creative Commons и не автором статьи. Поэтому если вы найдете какие-либо неточности, вы можете исправить их, обновив статью.

Молекулярные механизмы развития резистентности к инсулину



Всеволод Арсеньевич Ткачук
Александр Вячеславович Воротников
Опубликована Фев. 1, 2014
Последнее обновление статьи Ноя. 23, 2022
Эта статья опубликована под лицензией
")



Аннотация
Инсулиновая резистентность (ИР) ? это феномен, связанный с нарушением способности инсулина стимулировать захват глюкозы клетками-мишенями и снижать уровень глюкозы в крови. Ответное усиление секреции инсулина поджелудочной железой и гиперинсулинемия являются компенсаторными реакциями организма. Развитие ИР ведет к неспособности клеток-мишеней реагировать на инсулин, в результате чего развиваются сахарный диабет 2 типа (СД2) и метаболический синдром. По этой причине метаболический синдром на практике определяется как сочетание ИР с одной или несколькими патологиями, такими как СД2, артериальная гипертония, дислипидемия, абдоминальное ожирение, неалкогольная жировая болезнь печени и некоторые другие. Однако его физиологическим критерием всегда служит сочетание высокого уровня глюкозы и инсулина в крови.
ИР следует рассматривать как системный сбой эндокринной регуляции в организме. Физиологические причины ИР разнообразны. Основными являются пищевая перегрузка и накопление в клетках определенных липидов и их метаболитов, низкая физическая активность, хроническое воспаление и стресс различной природы, включая оксидативный и ?стресс эндоплазматического ретикулума? (нарушение распада поврежденных белков в клетке). Как показывают исследования последних лет, эти физиологические механизмы, скорее всего, реализуются по единому внутриклеточному сценарию. Им служит нарушение передачи сигнала от рецептора инсулина к его мишеням по механизму отрицательной обратной связи во внутриклеточных инсулин-зависимых сигнальных каскадах.
В данном обзоре рассмотрены физиологические и внутриклеточные механизмы действия инсулина; основное внимание уделено их нарушениям при развитии ИР. В заключении обсуждаются возможные направления ранней молекулярной диагностики и терапии ИР.
Ключевые слова
Сахарный диабет 2 типа, обратная связь, инсулиновая резистентность, инсулин-зависимая внутриклеточная сигнализация, фосфорилирование, белок IRS
СИСТЕМНОЕ ДЕЙСТВИЕ И ОРГАНЫ-МИШЕНИ ИНСУЛИНА
При поступлении пищи в организм концентрация глюкозы в крови повышается, что стимулирует секрецию инсулина β-клетками поджелудочной железы. Инсулин активирует поступление глюкозы в клетки скелетных мышц и жировой ткани. Эти клетки содержат много глюкозного транспортера 4 типа (ГЛЮТ4); инсулин вызывает его появление на клеточной поверхности, тем самым запуская транспорт глюкозы в клетки. В мышечных клетках (миоцитах) эта глюкоза запасается в виде гликогена, а в клетках жировой ткани (адипоцитах) она поступает в гликолиз, а из его продуктов синтезируются жиры. Инсулин действует и на гепатоциты – клетки основного метаболического органа – печени. В них он также стимулирует синтез гликогена и превращение глюкозы в липиды. В отличие от жировой ткани, печень не является депо жиров. Она активно экспортирует их в другие ткани, в том числе жировую, с помощью липопротеидных частиц.
Гепатоциты не имеют инсулин-зависимого переносчика ГЛЮТ4 и поэтому инсулин действует на них по механизму, отличному от такового в миоцитах и адипоцитах [1]. Он связан с изменением активности ферментов трех метаболических блоков, но не с транспортом глюкозы в клетки. Во-первых, инсулин ингибирует гликогенфосфорилазу, что тормозит распад и усиливает синтез гликогена в печени и мышцах. Во-вторых, он активирует ферменты гликолиза и ускоряет расщепление глюкозы до ацетил-кофермента А – субстрата для синтеза жирных кислот. Параллельно, инсулин инактивирует ферменты глюконеогенеза, тормозя обратный синтез глюкозы. В-третьих, инсулин снимает блок с ключевого фермента синтеза жирных кислот – ацетил-КоА-карбоксилазы, тем самым стимулируя образование малонил-КоА. В дополнение, инсулин подавляет активность липазы, расщепляющей триглицериды, способствуя их образованию из жирных кислот. Таким образом, суммарное действие инсулина на все ткани-мишени направлено на сдвиг метаболического равновесия в сторону превращения глюкозы в гликоген и липиды.
В промежутках между едой секреция инсулина снижается и снимается инсулиновый блок с глюконеогенеза и расщепления гликогена в печени. Инсулин-глюкагоновый индекс падает и начинает проявляться действие глюкагона и адреналина. Эти гормоны являются функциональными антагонистами инсулина, они усиливают распад гликогена, а глюкагон также стимулирует глюконеогенез и выход глюкозы из гепатоцитов.
При голодании синтез липидов в печени уменьшается, а в жировой ткани возрастает гидролиз триглицеридов. Его продукты – свободные жирные кислоты – транспортируются кровью в печень. Из-за необратимости реакций, катализируемых пируватдегидрогеназным комплексом, клетка не может превращать жирные кислоты в глюкозу. В качестве источника энергии из них преимущественно синтезируются кетоновые тела, которые транспортируются кровью к периферическим органам. Некоторые ткани, например миокард, используют кетоновые тела как основной источник энергии при голодании, но клетки мозга требуют глюкозы. Нужный для этого уровень глюкозы в крови поддерживается за счет глюконеогенеза, протекающего в печени; его источниками служат продукты катаболизма белков и аминокислот. Метаболический сдвиг в сторону кетоновых тел повышает интенсивность липолиза в жировой ткани и расход жировых запасов при длительном голодании.
Для организма важно сохранять баланс между поглощением, синтезом и выводом липидов из печени. Сдвиг этого баланса в сторону накопления липидов ведет к системной реакции, затрагивающей все инсулин-зависимые органы, и неминуемому развитию инсулинорезистентности (ИР).
Системная координация инсулин-зависимых тканей осуществляется центральной нервной системой совместно с гипоталамо-гипофизарной системой, играющей ключевую роль [2]. Эти органы также относят к инсулин-зависимым тканям – мишеням инсулина [3]. Они получают афферентные стимулы от желудка (грелин), поджелудочной железы (инсулин) и жировой ткани (лептин). Инсулин и лептин действуют в одном направлении, стимулируя запасание энергии в виде гликогена в печени и мышцах и триглицеридов в жировой ткани. При этом инсулин вызывает кратковременные, а лептин – долговременные реакции. Гормоны гипоталамуса (меланокортины, нейропептид Y, агутиподобный белок и другие) действуют на гипофиз, переводя афферентные стимулы в эфферентные [4]. За эфферентную регуляцию отвечают симпатическая и парасимпатическая нервные системы. Первая контролирует мобилизацию энергетических запасов, вторая – их накопление за счет вагусной регуляции секреторной активности β-клеток поджелудочной железы и периферических эффектов инсулина. Другие периферические гормоны также участвуют в системной координации инсулин-зависимых тканей. Наиболее значимые из них имеют адипоцитарное происхождение (адипонектин, резистин и другие адипокины) или являются местными провоспалительными цитокинами (TNF-α, интерлейкины-1 и -6, и другие). Именно с последними принято связывать роль воспаления как фактора риска и физиологического механизма ИР [5].
ФИЗИОЛОГИЧЕСКИЕ МЕХАНИЗМЫ РАЗВИТИЯ ИР
По своей природе ИР гетерогенна. Ее патофизиологической основой является системное нарушение взаимодействия четырех главных органов-мишеней инсулина. ИР возникает в этих органах последовательно и на первый взгляд независимо, но в итоге объединяет их все и приобретает системный характер [2]. Мышцы, печень и жировая ткань являются первичными органами-мишенями. В адипоцитах ИР развивается позже всех: жировая ткань часто сохраняет чувствительность к инсулину, в то время как печень и мышцы уже резистентны. Гипоталамус и гипофиз играют координирующую роль. Прямо или опосредованно, они также служат мишенью ИР. Таким образом, ИР – явление тканеспецифичное, и в его развитие могут вносить вклад несколько гормональных систем. На уровне организма ЦНС может играть ведущую и даже определяющую роль [6].
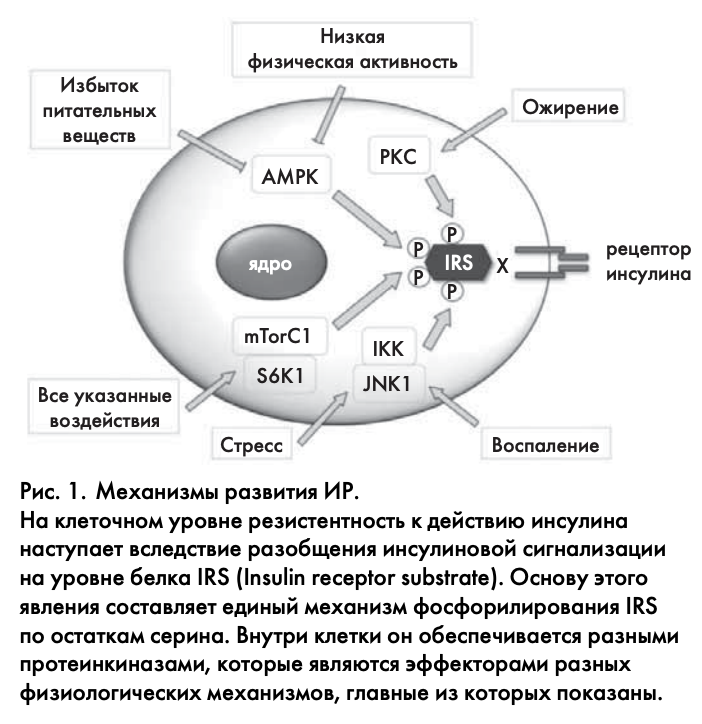
Вероятно, существует много причин возникновения и развития ИР, и все они пока не известны. На данный момент выделяют четыре основных физиологических механизма. Это избыточное питание и слабая физическая активность, ожирение, воспаление и стресс. Однако в конечном счете все они, видимо, реализуются в клетках по единому механизму фосфорилирования непосредственного субстрата инсулинового рецептора – белка IRS и нарушения его взаимодействия с рецептором (рис. 1). Поэтому используемое разделение на «физиологические» и «клеточные» механизмы довольно формально, и в первом случае отражает уровень всего организма, а во втором – не выходит за пределы клетки.
Избыточное питание и низкая физическая активность ведут к повышению в клетке уровня АТФ и снижению его метаболита – АМФ. В результате падает активность АМФ-зависимой протеинкиназы (АМРК). Этот фермент играет роль метаболического сенсора клетки, поддерживая энергетический гомеостаз [7]. О важности функций АМРК говорит тот факт, что она (или ее гомологи) присутствуют во всех эукариотических клетках от дрожжей до человека, включая клетки растений [8]. АМРК контролирует активность mTorC1 – первого белкового комплекса на основе киназы mTOR (mammalian target of rapamycin). Именно этот комплекс является главным переключателем между катаболизмом и анаболизмом в клетке [9]. В активном состоянии он стимулирует анаболические процессы, ведущие к синтезу белка, росту и делению клеток, синтезу жиров (липогенезу), а также к увеличению липидных депо организма за счет дифференцировки преадипоцитов в адипоциты (адипогенезу). Получая сигнал от АМРК, mTorC1 теряет активность; клетка переходит в режим катаболизма и утилизации запасов или поступающих источников энергии. По-видимому, этот переход как-то связан с реципрокной активацией второго белкового комплекса mTorC2. Именно mTorC2 отвечает за выход глюкозного транспортера ГЛЮТ4 на клеточную мембрану и инсулин-зависимый транспорт глюкозы внутрь адипоцитов и миоцитов [10].
При физической нагрузке идет интенсивный гидролиз АТФ в миоцитах. Фермент аденилаткиназа регенерирует АТФ в реакции 2 АДФ > АТФ + АМФ, а образуемый АМФ активирует АМРК. Та выключает mTORC1 и запускает утилизацию энергетических запасов. Таким образом, система АМРК является также сенсором физической нагрузки. В отсутствие последней активность АМРК снижается, а mTORC1 – возрастает. Вместе эти события являются физиологически скоординированной реакцией клеток на избыточное питание и отсутствие физической нагрузки.
АМРК является также мишенью адипонектина – гормона жировой ткани [11]. Адипонектин повышает чувствительность к инсулину, но его секреция у тучных людей снижена [12]. Это приводит к тому, что при ожирении АМРК ингибирована, а mTOR активен. Кроме того, адипонектин препятствует развитию ИР, активируя распад церамидов и накопление в клетке важной сигнальной молекулы – сфингозин-1-фосфата [13]. В условиях ожирения и недостаточности адипонектина распад церамида нарушен и включается церамид-зависимый механизм индукции ИР, не задействующий АМРК и mTOR.
Гиперлипидемия (ожирение) тесно связана с резистентностью к инсулину. Хотя до конца неясно, какое из этих явлений первично, многие клинические данные указывают на ожирение как причину ИР [14]. В свою очередь, ИР ускоряет дальнейший набор веса и способствует ожирению [2], действуя по механизму положительной обратной связи, детали которого пока неизвестны. Ограничительная диета и потеря веса восстанавливают чувствительность к инсулину у людей c малоподвижным образом жизни [15].
Патологическое ожирение, связанное с ИР, имеет характерную особенность. Этот жир имеет висцеральное (абдоминальное) расположение, т.е. откладывается в клетках печени, мышц, сердца, сосудистой стенки и других органов, концентрируясь в области брюшины. Напротив, в норме жиры депонируются в адипоцитах, и эта жировая прослойка располагается подкожно и гораздо более равномерно распределяется в организме. В связи с этим, патологические жировые отложения принято называть эктопическими, тем самым обозначая ненормальность их расположения в организме [16]. Установлено, что появление именно эктопического, но не висцерального жира ведет к развитию ИР и метаболическим изменениям [17].
Эктопическое распределение жиров не следует путать с их накоплением вне клеток. Результаты, полученные на животных моделях и с пациентами, показывают, что степень выраженности ИР коррелирует с накоплением жиров внутри клеток, а не в межклеточном пространстве [1]. Именно внутриклеточные липиды нарушают передачу сигнала от рецептора инсулина и снижают инсулин-зависимый захват глюкозы в клетках нежировых тканей, вызывая развитие ИР и СД2 [16]. Этот физиологический механизм согласуется с представлениями о том, что ИР развивается раньше в печени и скелетных мышцах [2], тогда как жировая ткань какое-то время остается инсулин-чувствительной [18]. Скорее всего, ИР проявляется прежде всего в печени, и лишь затем развивается в других органах, причем с разной временной задержкой [19]. Длительное эктопическое накопление жиров в печени (ожирение) ведет к развитию неалкогольной жировой болезни печени [20].
Липиды составляют широкий спектр молекул с различной структурой и функциями. При попадании в клетку свободные жирные кислоты быстро подвергаются тиолированию с образованием ацил-КоА. В печени ацил-КоА подвергается β-окислению, а в адипоцитах используется для синтеза триглицеридов. Кроме того, он используется во всех клетках для этерификации сфингозина до церамидов. Некоторые липидные метаболиты (например, диацилглицериды и церамиды) рассматриваются как вторичные посредники в различных сигнальных каскадах клетки. Они представляют собой основные липиды, повышение уровня которых инициирует ИР.
В последние годы в общих чертах выяснен механизм развития ИР в печени [1]. Он связан с активацией «новых» изоформ протеинкиназы С (РКС) липидными метаболитами, но не ионами Са2+, как этого дополнительно требуют типичные изоформы РКС [21]. Прямо или опосредованно, «новые» изоформы РКС нарушают сигнализацию от рецептора инсулина внутрь клетки (см. ниже). Эктопическое накопление липидов в гепатоцитах повышает уровень диацилглицеридов, которые и являются активаторами «новых» РКС. Содержание диацилглицеридов в липидных каплях в цитоплазме гепатоцитов тучных людей четко коррелирует c активностью PKCε и со степенью ИР [22].
Механизм развития ИР в миоцитах, по-видимому, аналогичен. Он также задействует «новые» изоформы РКС и нарушение инсулиновой сигнализации в клетках [23, 24]. Динамика ИР в мышечной ткани совпадает с накоплением диацилглицерина в миоцитах, ухудшением передачи сигнала от инсулина и снижением захвата глюкозы мышечными клетками [25]. Уровень эктопических липидов в мышечной ткани является более надежным предиктором ИР, чем уровень циркулирующих в кровотоке жирных кислот [26].
Следует особо отметить, что липидный механизм с участием РКС является главным, если не единственным, обеспечивающим развитие ИР в печени у людей. Специальный сравнительный анализ фактически не выявил вклада таких альтернатив, как воспаление или стресс эндоплазматического ретикулума [22]. Вместе с тем, была обнаружена обратная корреляция между ИР в печени и уровнем адипонектина в плазме крови пациентов [22], что говорит о вероятной роли адипоцитарных гормонов в инициации ИР. Эти данные отличаются от результатов исследований на животных моделях, согласно которым вклад воспаления и эндоплазматической реакции в развитие ИР значителен [1]. Кроме того, экспериментальные данные указывают на то, что в мышцах грызунов развитие ИР связано с нарушением функции митохондрий [23]. Хотя ни детали этого механизма, ни его наличие у людей остаются неясными, можно предположить, что в нем задействованы свободные радикалы, как это часто бывает при митохондриальных дисфункциях.
Роль РКС в развитии ИР в жировой ткани не продемонстрирована. Особенности физиологии этой ткани позволяют предполагать, что там реализуются иные механизмы. В отличие от миоцитов и гепатоцитов, адипоциты в норме содержат много триглицеридов и динамично меняют их уровень и состав. Как следствие, они постоянно имеют высокий уровень диацилглицеридов, который значительно превышает тот, что необходим для полной активации любых РКС. Поэтому механизм развития ИР в этой ткани пока неясен. Возможно, он задействует воспаление, стрессовые реакции или дисфункцию митохондрий. Однако следует помнить, что ИР в этой ткани возникает как продолжение патологических изменений в печени и мышцах.
Воспаление. Эмпирические наблюдения ИР у пациентов с сепсисом [27, 28] и уровня цитокинов у людей с ожирением и диабетом [29, 30] позволили предположить, что причиной развития ИР может быть аномальная активация врожденного иммунитета. Эксперименты на животных показали повышенный уровень TNFα в жировой ткани при ожирении; его нейтрализация восстанавливала захват глюкозы периферическими тканями [31]. Эти результаты были подтверждены и у людей, где уровень TNFα коррелировал с ИР и падал при снижении массы тела [32]. Наконец, был продемонстрирован механизм, согласно которому воспалительные цитокины могут прерывать сигнализацию от инсулинового рецептора внутри [33]. Все эти данные послужили основой для создания концепции о связи между воспалением и ИР [5].
Воспаление вызывает активацию определенных сигнальных каскадов, и это является частью нормальной физиологической реакции клетки не только при наличии патологий. Например, и у здоровых людей, и у пациентов с ожирением, но без диабета, физическая нагрузка в аэробных условиях стимулирует экспрессию цитокинов в скелетных мышцах (MCP1 и IL-6) и ведет к активации сигнального каскада с участием NFκB. Однако у пациентов с СД активность этого каскада исходно высокая и существенно не повышается при физической нагрузке [34].
При голодании или дисфункции в адипоцитах аномально усиливаются липолиз и продукция хемокинов [35]. Хемокины вызывают движение в жировую ткань макрофагов, где эти клетки активируются в активированные макрофаги. Они передают информацию о воспалении другим клеткам с помощью усиленной секреции цитокинов, в том числе TNFα и IL-6. Действуя на адипоциты, эти цитокины дополнительно стимулируют в них липолиз. Так, инфузия IL-6 здоровым мужчинам усиливала липолиз и окисление липидов в пределах бедренного сосудистого бассейна [36]. Механизм действия цитокинов на липидный обмен в адипоцитах отчасти связан с подавлением экспрессии белков, стабилизирующих липидные капли – перилипина [37] и FSP27 [38].
Трансгенные мыши с повышенной продукцией хемокина CCL2 в адипоцитах имеют значительно больше активированных макрофагов в жировой ткани [39]. У таких животных развивается периферическая и печеночная ИР, причем последняя ведет к стеатозу. Блокада CCL2 в условиях высококалорийной диеты защищает этих мышей от развития ИР и ведет к снижению уровня липидов в печени [40]. В мышечных клетках цитокины также стимулируют окисление липидов, а в экстремальных ситуациях могут даже запускать протеолиз белков и атрофию мышечной ткани.
Напротив, в печени цитокины оказывают противоположное действие, тормозя окисление липидов и усиливая липогенез [5]. Это значит, что в принципе активированные макрофаги могут влиять на межтканевой энергетический баланс, сдвигая синтез липидов из жировой ткани в печень. Вряд ли можно ожидать, что этот эффект успевает значительно развиться при кратковременном или локальном воспалении. Однако при системном и хроническом воспалении активация макрофагов имеет массовый и длительный характер. В таких условиях сильно повышается вероятность системных изменений энергетического баланса, что может приводить к перераспределению липидов и их эктопическому накоплению в печени. Экспериментальные результаты согласуются с участием активированных макрофагов в инициации эктопического накопления липидов и ИР, по крайней мере в животных моделях. Насколько этот физиологический механизм реализуется у человека, остается во многом неясным.
Механизм деградации поврежденных белков (так называемый UPR, англ. Unfolded protein response) стимулирует адаптивные реакции клеток в условиях недостатка пищевых ресурсов [41]. Он запускается тогда, когда по каким-либо причинам истощается емкость внутриклеточной системы фолдинга вновь синтезируемых белков. Эта система располагается на эндоплазматическом ретикулуме (ЭР) клетки, в связи с чем UPR часто считают реакцией клетки на стресс, связанный с ЭР. Из-за этого UPR иногда называют «стрессом ЭР», хотя более верно считать UPR следствием стресса ЭР [42]. Хронический стресс ЭР и активация UPR ведут к оксидативному стрессу и образованию свободнорадикальных молекул [43]. Таким образом, оксидативный стресс является составной частью стресса ЭР, а эти два механизма сильно перекрываются на молекулярном уровне.
Все внутриклеточные системы активации UPR чувствительны к уровню глюкозы и запускаются при его повышении. Это значит, что высококалорийная углеводная диета ведет к активации UPR. Физиологический смысл этой реакции заключается в защите от углеводной перегрузки и быстром переключении метаболизма клетки на синтез жиров (липогенез). В этом смысле UPR почти дублирует действие инсулина на метаболический баланс клетки, не влияя только на транспорт глюкозы. Способность UPR вызывать развитие ИР может в конечном итоге зависеть от того, сдвигает ли UPR баланс в сторону липогенеза так, что происходит эктопическое накопление липидов.
В настоящее время система UPR рассматривается не просто как один из активаторов ряда метаболических реакций клетки, а как жизненно важный интегратор протекающих в ней анаболических и катаболических процессов [41]. По своей значимости систему UPR можно сравнивать с такими мастер-регуляторами клеточного метаболизма, как комплексы mTOR и АМРК. Не случайно и не удивительно, что эти три системы тесно взаимосвязаны в клетке, обрабатывая сходные сигналы и влияя на активность друг друга [44]. Как следствие, система UPR не только реагирует на метаболические сигналы, но и дает клетке дополнительный способ восприятия системных изменений, в том числе характерных для диабета и метаболического синдрома. К ним относятся ожирение, воспаление и стресс разной этиологии. Поэтому UPR задействует те же сигнальные молекулы при развитии ИР, что и другие, упомянутые выше физиологические механизмы [45].
На первый взгляд кажется нелогичным, что UPR, активируемый глюкозой, нарушает передачу сигнала от инсулинового рецептора [45], а снижение стресса ЭР усиливает инсулиновую сигнализацию [46]. Однако это явление следует рассматривать в контексте обратной регуляции, столь характерной для метаболического контроля. ИР развивается как защитная реакция клетки и организма в целом на пищевую перегрузку, ожирение, воспаление и стресс, а UPR служит одним из путей ее реализации. В конечном счете, активация UPR ведет к выключению инсулиновой сигнализации на том же уровне белка IRS, как это происходит в случае mTOR-зависимого, липид-зависимого и воспалительного механизмов.
ИНСУЛИНОВАЯ СИГНАЛИЗАЦИЯ
В большинстве клеток рецептор инсулина служит главным образом для запуска PI3-киназного каскада (рис. 2) [47, 48]. Его главной мишенью в клетке служит протеинкиназа В, более известная как Akt [49]. Как и все рецепторные тирозинкиназы, рецептор инсулина активирует также несколько других сигнальных систем. Отличительной чертой инсулиновой сигнализации является участие каркасного белка IRS – субстрата инсулинового рецептора. Как отражает название, IRS выступает главным (если не единственным) партнером активированного рецептора. Он не имеет ферментативной активности и служит местом посадки для ряда сигнальных молекул-мишеней рецептора инсулина. Таким образом, IRS в прямом смысле выполняет «передаточную» функцию, и без него сигнал от рецептора в клетку не проходит.
Мембранные взаимодействия являются второй характерной чертой инсулиновой сигнализации. Они обеспечиваются наличием в белках-участниках специальных структур (РН-, РХ и FYVE-доменов), которые связывают фосфатидилинозитолы – мембранные фосфолипиды, выполняющие сигнальные функции. IRS содержит на N-конце РН-домен плекстриновой гомологии, которым заякоревается на мембране вблизи от рецептора. При связывании инсулина рецептор фосфорилирует себя по остаткам тирозина, и эти остатки узнаются фосфотирозин-связывающим (PTB) доменом, расположенным на С-конце белка IRS. Далее рецептор фосфорилирует IRS по С-концевым остаткам тирозина, после чего те связываются сSH2- и PTB-доменами приходящих из цитозоля эффекторных белков. Такими белками являются PI3-киназа, фосфотирозиновая фосфатаза 2 (SHP-2) и адаптерные белки Shc или Grb2. Последние активируют малую ГТФ-азу Ras и Erk-каскад МАР-киназ, который запускает деление клеток и отличается от стресс-зависимого каскада JNK-МАР-киназ.
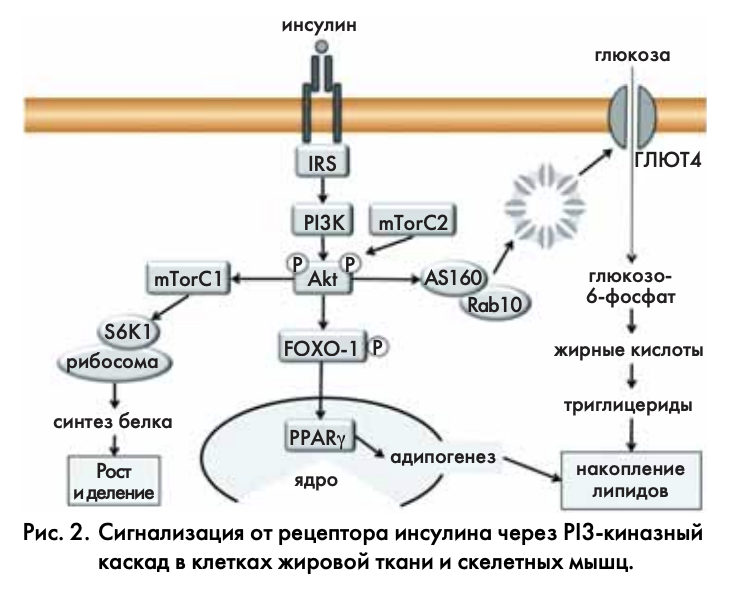
Активированная связыванием с IRS, PI3-киназа фосфорилирует третье положение инозитольного кольца фосфатидилинозитолбисфосфата (РIP2), образуя PIP3 (отсюда и название фермента – фосфатидилинозитол-3-киназа). Это событие имеет принципиальное значение, так как большинство РН-доменов узнает фосфат именно в 3-м положении инозитольного кольца. Такой РН-домен есть в Akt; за счет него Akt рекрутируется на мембрану. Аналогичный домен есть и в фосфоинозитид-зависимой протеинкиназе PDK1, которая также связывается с мембраной и фосфорилирует Akt по остатку Thr308 внутри активационной петли. Так происходит рецептор-зависимая активация Akt [47].
Дополнительный уровень сложности в PI3-киназном каскаде возникает потому, что одного фосфорилирования Thr308 недостаточно для того, чтобы Akt стала активной. Для этого требуется еще одно фосфорилирование по остатку Thr473 (рис. 2). Долгое время оно оставалось загадкой в отношении своей значимости и фермента, его катализирующего. Лишь относительно недавно стало понятно, что таким ферментом служит киназа mTOR в составе второго белкового комплекса mTORC2, а само это фосфорилирование является необходимым, но не достаточным условием для активации Akt. Оно протекает в фоновом режиме, но без него фосфорилирование по Thr308 эффекта не имеет [48, 50]. Другими словами, комплекс mTORC2 «праймирует» Akt к активации инсулином в контексте PI3-киназного каскада. Учитывая, что Akt активирует первый комплекс mTORC1 и таким образом запускает синтез белка, адипогенез, а также механизм выживания клеток в неблагоприятных условиях [49], регуляторный контроль внутри этого сигнального модуля становится крайне запутанным и очень сложно интерпретируемым [51]. На данный момент многие вопросы остаются открытыми и ситуация еще больше усложняется наличием многочисленных обратных связей и центральной ролью mTOR в регуляции метаболизма клетки [52].
Активированная Akt фосфорилирует более 100 субстратов и регулирует почти все жизненно важные функции клетки – метаболизм, рост, движение, деление, выживание и клеточную смерть [49]. Действуя через mTORC1, Akt активирует рибосомальную S6-киназу (S6K1) и выключает ингибитор фактора инициации трансляции эукариот 4E-BP. Таким образом, mTorC1 стимулирует синтез белка, рост и деление клеток (рис. 2). mTorC1 участвует и в регуляции липидного обмена и гомеостаза холестерина, контролируя фосфорилирование транскрипционных факторов SREBP1 и PPARγ, а также липина-1, регулирующего поступление триглицеридов в жировые капли адипоцитов [52].
Akt фосфорилирует и активирует AS160 – фактор обмена гуаниловых нуклеотидов малых ГТФаз семейства Rab (рис. 2) [10]. Эти белки регулируют внутриклеточный транспорт везикул, а Rab10 отвечает за слияние ГЛЮТ4-содержащих везикул с мембраной клетки [53]. Таким путем Akt запускает экспонирование глюкозного транспортера и транспорт глюкозы в адипоциты и миоциты. В миоцитах глюкоза фосфорилируется и направляется на синтез гликогена, тогда как в адипоцитах она служит для липогенеза и из нее синтезируются триацилглицериды.
Akt также фосфорилирует транскрипционный фактор FOXO1, который активирует PPAR-γ – мастер-регулятор адипоцитарной дифференцировки (рис. 2). Так, Akt запускает адипогенез и увеличивает число адипоцитов в жировой ткани [54]. Таким образом, действуя через Akt, инсулин стимулирует адипогенез и липогенез, приводя к накоплению липидов в клетках. Чтобы избежать гиперактивации каскада и эктопического накопления липидов клетки, используют механизмы обратной связи, направленные на временное разобщение рецептора и сигнал-проводящей цепочки. В следующем разделе обсуждается, как постоянная активация этой обратной связи ведет к развитию ИР.
ВНУТРИКЛЕТОЧНЫЙ МЕХАНИЗМ РАЗВИТИЯ ИР
Внутриклеточной основой ИР считается нарушение сигнализации от инсулинового рецептора, что разобщает действие инсулина и соответствующую реакцию клеток. Сейчас практически не вызывает сомнений, что это разобщение происходит на уровне субстрата инсулинового рецептора – белка IRS. Для инсулин-резистентных клеток характерно повышенное фосфорилирование IRS по остаткам серина. Оно препятствует тирозиновому фосфорилированию IRS, нужному для проведения сигнала от рецептора внутрь клетки. Таким образом, развитие ИР сопряжено с сериновым фосфорилированием белка IRS.
Как отмечено выше, физиологические причины ИР разнообразны, ее ткани-мишени (жировая ткань, мышцы и печень) имеют различную физиологию и время развития ИР. В силу этих причин, в разных клетках разными способами запускается единый механизм разобщения инсулиновой сигнализации путем серинового фосфорилирования белка IRS, имеющий свои тканеспецифичные особенности [55]. Во-первых, разные физиологические механизмы активируют в клетке разные сигнальные каскады, которые одинаково замыкаются на сериновом фосфорилировании IRS (см. рис. 1). Во-вторых, за это фосфорилирование отвечают разные протеинкиназы, но каждая из них фосфорилирует в IRS один или несколько вполне определенных остатков. В-третьих, IRS имеет много сериновых остатков, нарушающих передачу инсулинового сигнала. В-четвертых, каждая ткань-мишень инсулина имеет свой набор фосфорилируемых остатков; он определяется физиологическими особенностями и механизмами развития ИР в данной ткани.
Теоретически, распределение фосфорилируемых остатков в IRS и задействованные протеинкиназы позволяют судить, какие внутриклеточные каскады опосредуют развитие ИР. В свою очередь, зная эти каскады, можно определить физиологический механизм развития и, по сути, исходную причину ИР. Например, инсулин и инсулиноподобный фактор роста (IGF-1), так же как и аминокислоты, действуют через инсулин-зависимый сигнальный каскад с участием Akt и комплекса mTORC1, вызывая фосфорилирование Ser302 и Ser632/635 в IRS мышей и, соответственно, Ser307 и Ser636/639 в IRS человека [23]. В то же время, интерлейкины и TNFα опосредуют воспалительные эффекты и вызывают фосфорилирование Ser307 в клетках мышей и Ser312 у человека. Активация разных изоформ РКС (δ, ε, θ или ζ) происходит при дислипидемии и эктопическом ожирении, даже если последнее визуально не проявляется. Каждая изоформа РКС вызывает фосфорилирование своего набора остатков в IRS, однако преимущественно модифицируется Ser1101 (в IRS человека). Эта особенность механизмов возникновения и развития ИР делает принципиально возможным определение этиологии ИР и проведение ее ранней диагностики по профилю фосфорилирования одного белка, которым является IRS. Исследования последних лет подтверждают ключевую роль фосфорилирования IRS в развитии ИР и существенно детализируют «молекулярный профиль» IRS [55].
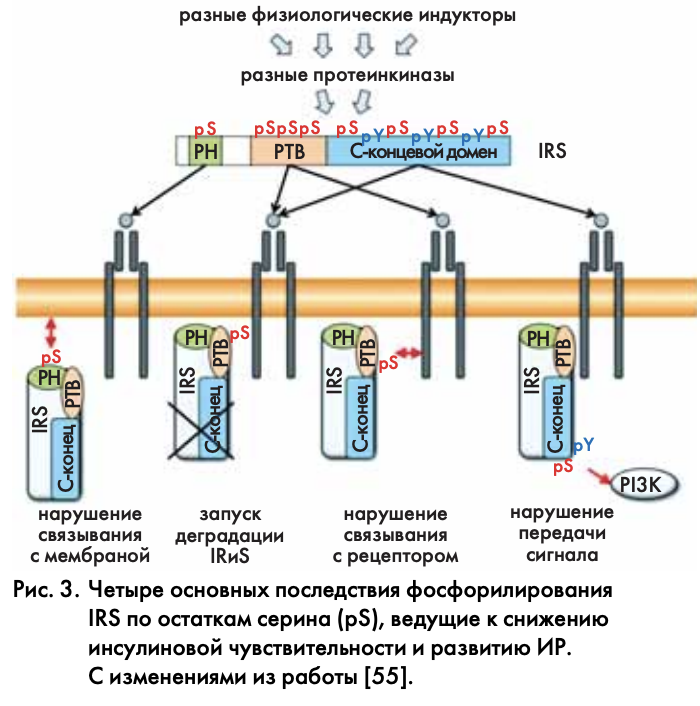
На молекулярном уровне фосфорилирование IRS по остаткам серина имеет четыре последствия, которые разными способами нарушают передаточную функцию этого белка (рис. 3). В первом случае фосфорилирование нарушает связывание PH домена IRS с мембраной, уводя IRS от рецептора. Во втором и третьем случаях фосфорилирование внутри PTB домена или рядом с ним вызывает или диссоциацию IRS от рецептора, или деградацию IRS. Наконец, фосфорилирование внутри C-концевого домена нарушает связывание IRS с эффекторами и передачу сигнала внутрь клетки [53, 55]. Однако в любом случае разные физиологические индукторы и механизмы задействуют внутри клеток разные киназы, вызывая фосфорилирование IRS по разным остаткам. Эти внутриклеточные механизмы суммированы на рис. 4 и ниже разбираются более подробно.
Избыточное питание и низкая физическая нагрузка снижают активность АМРК и, как следствие, активируют mTORC1. Одной из главных мишеней mTORC1 является рибосомальная киназа S6K1; в ее задачу входит фосфорилирование S6-компонента малой субъединицы рибосомы и запуск синтеза белка (трансляции). Однако помимо этого S6K1 фосфорилирует IRS1 по нескольким сериновым остаткам, прерывая его передающие функции [52, 56]. Этот внутриклеточный механизм обратной связи в инсулиновой сигнализации (см. рис. 4) считается ключевым для развития ИР, ожирения и диабета 2 типа; он также играет важную роль в опухолевой прогрессии [57].
Есть несколько возможностей инсулин-независимой активации Akt/mTORC1, при которых интенсивно работает обратная связь и фосфорилирование IRS поддерживается даже при нарушенной передаче сигнала от рецептора к Akt (рис. 4). Во-первых, возможна альтернативная активация Akt за счет фосфорилирования остатка Ser473 в ее гидрофобном домене. Эту реакцию катализирует та же киназа mTOR, находясь в составе второго комплекса (mTORC2) [48]. Механизм активации mTORC2 окончательно не ясен, но известно, что его запускают рецепторы факторов роста. Они стимулируют образование внутриклеточных активных форм кислорода (АФК), которые усиливают фосфорилирование [58] и активацию Akt [59].
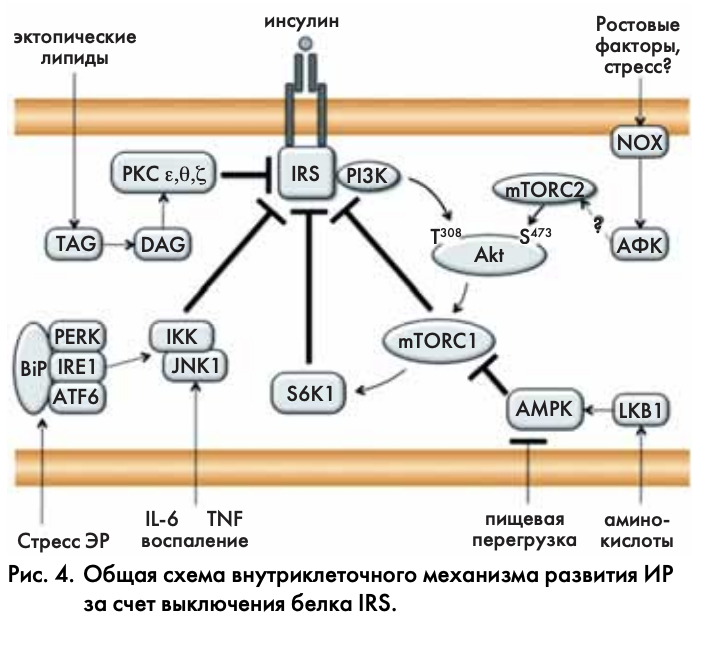
Другим путем инсулин-независимой активации mTORC1 служит выключение его ингибитора – киназы АМРК. Это происходит при пищевой перегрузке, накоплении в клетке АТФ и соответствующем снижении АМФ [7]. Активация mTORC1 происходит также при воспалении [41] и стрессе ЭР [44]. Таким образом, почти все физиологические механизмы развития ИР в той или иной степени замыкаются на активации mTORC1 и его непосредственной мишени – S6K1. mTORC1 и S6K1 прямо фосфорилируют набор сериновых остатков в белке IRS (см. рис. 3), нарушая передачу инсулинового сигнала.
Мыши, нокаутные по гену S6K1, имеют повышенную чувствительность к инсулину и менее склонны к ожирению, связанному с возрастом или вызванному жирной диетой [56]. Пока остается неясным, в какой ткани реализуется этот механизм у человека. Учитывая данные, полученные от пациентов с неалкогольной болезнью печени [22], можно считать, что у людей этот механизм по большей части характерен для жировой ткани, так как в печени и мышцах преимущественно работает липид-зависимый механизм с участием РКС.
Липид-зависимый механизм получил весомое экспериментальное обоснование. Он постулирует, что в клетках печени и скелетных мышц за выключение IRS отвечает протеинкиназа С (РКС). При этом сама РКС не фосфорилирует IRS1/2, а лишь только инициирует каскад обратной связи, ведущий к серин-треониновому фосфорилированию IRS1/2 [1]. В мышцах эту функцию выполняет РКС-θ, а в печени – РКС-ε. Обе изоформы относятся к группе «новых» РКС, которые не требуют ионов Са2+ для активации, но значительно более чувствительны к диацилглицеролу, чем классические изоформы РКС. Эти свойства могут, по крайней мере отчасти, объяснить связь ИР с ожирением. «Новые» изоформы PKC вызывают ИР в печени и мышцах без участия инсулина и его рецептора, поскольку активируются продуктами эктопических липидов независимо от инсулина (рис. 4). Есть также данные о том, что ту же функцию выполняет РКС-ζ [60]. Однако ее связь с ожирением остается неясной, так как РКС-ζ относится к «нетипичным» изоформам РКС и не использует Са2+ и диацилглицерол в качестве активаторов [21]. Согласно другой точке зрения, РКС-ζ участвует в церамид-зависимом механизме выключения IRS (см. ниже).
Липид-зависимый механизм получил подтверждение и в животных моделях. Мыши, у которых отсутствует PKC-θ, не развивали быстрой ИР при массивной инфузии липидов [61]. Аналогично, мыши, нокаутные по PKCε, не развивали ИР в условиях высококалорийной диеты, хотя уровень липидов в печени повышался [62]. PKCδ – это еще одна изоформа PKC, связанная с развитием ИР в печени. Выключение PKCδ ведет к снижению уровня липидов в печени и повышению чувствительности к глюкозе, тогда как сверхэкспрессия PKCδ вызывает развитие ИР и стеатоза печени [63, 64].
Церамид-зависимый механизм является исключением из общего правила, так как направлен не на IRS, а на ингибирование Akt. Считается, что синтез церамидов запускает киназа IKK в контексте сигнального каскада NF-κB, активируемого Toll-подобным рецептором 4 типа (TLR4) [65] или рецептором TNFα. Таким образом, образование церамидов может быть частью воспалительного ответа клетки. Некоторые исследователи полагают, что насыщенные жирные кислоты выступают лигандами TLR4, вызывая развитие ИР в условиях гиперлипидемии [66]. Церамиды действуют на активность Akt2 двумя способами. Один связан с активацией PKCζ, которая затем взаимодействует с Akt и выводит ее из инсулинового каскада [67]. В другом варианте церамиды активируют фосфатазу РР2А, которая дефосфорилирует и инактивирует Akt [68]. В любом случае церамиды снижают активность Akt, нарушая передачу сигнала от рецептора инсулина. Следует заметить, что церамид-зависимый механизм был обнаружен и исследован в клеточной модели ИР, но в животных моделях пока не подтвержден.
Воспаление почти всегда связано с активацией двух внутриклеточных сигнальных каскадов; они опосредуют воспалительную реакцию и фосфорилирование IRS1 (рис. 4). Каскад NF-κB связан с активацией киназы IKK, которая прямо фосфорилирует IRS или действует опосредованно, запуская синтез церамидов (см. выше). Другой каскад запускает стресс-зависимую МАР-киназу JNK1; она прямо фосфорилирует IRS [33].
Мыши, нокаутированные по ε-изоформе IKK, имеют повышенный катаболический статус, существенно сниженный объем жировой ткани и не развивают ИР [69]. Аналогичный фенотип имеют мыши, дефицитные по JNK1 [70]. Нокаут гена JNK1 в жировой ткани защищает мышей от развития стеатоза и печеночной формы ИР, хотя они нормально набирают вес и объем жировой ткани [71]. У этих мышей отмечено повышенное воспаление в жировой ткани и умеренное развитие стеатоза, хотя чувствительность к инсулину в печени и жировой ткани не изменяется.
Стресс ЭР (механизм UPR) запускается при перегрузке ЭР синтезированными или неправильно свернутыми белками. Он опосредован тремя сигнальными модулями (рис. 4), физически связанными с мембраной ЭР [41, 42]. Во-первых, это PERK-киназа, которая фосфорилирует и ингибирует фактор инициации трансляции 2a эукариот, подавляя синтез белка. Во-вторых, это инозитол-зависимая киназа IRE1, имеющая эндорибонуклеазную активность. Она активирует нетрадиционный сплайсинг мРНК фактора транскрипции ХВР1 и его трансляцию [44]; в свою очередь, ХВР1 запускает экспрессию шаперонов и белков протеосом, нужных для преодоления стресса ЭР. В-третьих, это фактор транскрипции ATF6; он активируется путем частичного протео лиза, и его цитозольный фрагмент перемещается в ядро, где запускает экспрессию шаперонов ЭР. PERK, IRE1 и ATF6 образуют неактивный комплекс с шапероном BiP/GRP78 (HSP70 или HSPA5). BiP служит метаболическим сенсором системы UPR; его экспрессия усиливается при глюкозном голодании. При активации UPR, BiP уходит из комплексов с PERK, IRE1 и ATF6, что ведет к активации этих сигнальных модулей. Конечной мишенью по крайней мере одного из них (IRE1) является МАР-киназа JNK1, фосфорилирующая IRS [45,72]. Таким образом, стресс-зависимый и воспалительный пути используют одного исполнителя – JNK1, вызывая развитие ИР.
Оксидативный стресс и дисфункция митохондрий ведут к повышенной продукции свободнорадикальных молекул и, в частности активных форм кислорода (АФК). С одной стороны, АФК активируют стресс ЭР [44], по-видимому, PERK-зависимым образом [73]. Как отмечено выше, в результате активируется JNK1, происходит сериновое фосфорилирование IRS и инсулиновый каскад нарушается. С другой стороны, АФК поддерживают инсулиновую сигнализацию и повышают чувствительность к инсулину в адипоцитах [59]. Эти противоречивые данные позволили говорить о так называемом «редокс-парадоксе» в инсулиновой сигнализации [74].
Редокс-парадокс разрешается, если принять во внимание разные источники и значительно более высокий внутриклеточный уровень АФК при оксидативном стрессе. В случае стресса избыток АФК запускает JNK1-зависимый механизм, характерный для воспаления и стресса ЭР. Это ведет к сериновому фосфорилированию IRS, разобщению инсулиновой сигнализации и снижению активации Akt. Напротив, в отсутствие стресса, физиологическая активация рецепторов факторов роста стимулирует сборку НАДФН-оксидазных комплексов (NOX) и регулируемую продукцию АФК (рис. 4). Эти АФК работают в малых количествах и локально. Действуя как вторичные посредники, они усиливают передачу сигнала от рецепторов внутрь клетки [75]. Таким путем они обеспечивают полноценное фосфорилирование и активацию Akt в адипоцитах [58, 59]. Ближайшие исследования должны показать, связано ли действие АФК с редокс-зависимой активацией mTORC2 (см. рис. 4).
Перспективы молекулярной диагностики и терапии ИР
В последние годы достигнут значительный прогресс в понимании механизмов развития резистентности к инсулину в тканях-мишенях. Скорее всего, первой мишенью ИР является печень. Накопление эктопических липидов в гепатоцитах стимулирует быстрое развитие патологии в мышцах, после чего изменения постепенно приходят в жировую ткань. Гипоталамо-гипофизарная система координирует эти процессы и может участвовать в них с самых первых этапов. Высококалорийное питание и недостаток физической активности, а также наличие стресса и воспаления существенно усугубляют патогенез ИР, в конечном итоге приводя к развитию метаболического синдрома, СД, гепатоцеллюлярных и сердечно-сосудистых патологий.
Принципиальным знанием последних лет стал единый молекулярный механизм развития инсулиновой резистентности, связанный с сериновым фосфорилированием IRS. Это значит, что повышение уровня фосфорилирования IRS может служить индикатором и методом ранней диагностики ИР. Более того, расположение фосфорилируемых остатков в молекуле IRS может указывать на физиологические причины возникновения ИР и сделать возможным подбор соответствующей терапии для данного пациента. Дальнейший прогресс в этой области, несомненно, будет связан с разработкой новых лекарственных препаратов, действие которых направлено на активность внутриклеточных ферментов, прямо или косвенно обеспечивающих фосфорилирование IRS. Это совершенно новая область фармакологии, уходящая на более глубокий клеточный уровень, чем современные подходы, традиционно направленные на гормон-рецепторные взаимодействия снаружи клеток. Она, безусловно, требует тщательного решения многих задач как прикладного, так и фундаментального характера, включая адресную доставку и специфичность фармакологических агентов.
В рамках индивидуальной медицины перспективна возможность «молекулярного профилирования» IRS в тканевых биоптатах. Она обеспечена современными средствами высокопроизводительного анализа, такими как масс-спектрометрия, и позволяет выяснить набор остатков, фосфорилируемых в IRS у людей с метаболическим синдромом или с диабетом. Эти знания дают информацию о наличии и этиологии ИР у данного пациента, а также позволяют осуществлять дифференциальную диагностику сопутствующих заболеваний и постоянно корректировать наши текущие представления о молекулярных механизмах развития ИР.
СПИСОК ЛИТЕРАТУРЫ
- Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. The Lancet 2010;375(9733):2267-2277. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0140673610604084https://doi.org/10.1016/S0140-6736(10)60408-4.
- Isganaitis E, Lustig RH. Fast Food, Central Nervous System Insulin Resistance, and Obesity. Arteriosclerosis, Thrombosis, and Vascular Biology 2005;25(12):2451-2462. Available from: http://atvb.ahajournals.org/cgi/doi/10.1161/01.ATV.0000186208.06964.91PubMed PMID:16166564.https://doi.org/10.1161/01.ATV.0000186208.06964.91.
- Grayson BE, Seeley RJ, Sandoval DA. Wired on sugar: the role of the CNS in the regulation of glucose homeostasis. Nat Rev Neurosci 2013;14(1):24-37. Available from: http://www.nature.com/doifinder/10.1038/nrn3409PubMed PMID:23232606.https://doi.org/10.1038/nrn3409.
- Flier J. Obesity WarsMolecular Progress Confronts an Expanding Epidemic. Cell 2004;116(2):337-350. Available from: http://linkinghub.elsevier.com/retrieve/pii/S009286740301081XPubMed PMID:14744442.https://doi.org/10.1016/S0092-8674(03)01081-X.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444(7121):860-867. Available from: http://www.nature.com/doifinder/10.1038/nature05485PubMed PMID:17167474.https://doi.org/10.1038/nature05485.
- Schwartz MW, Seeley RJ, Tschöp MH, Woods SC, Morton GJ, Myers MG, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013;503(7474):59-66. Available from: http://www.nature.com/doifinder/10.1038/nature12709PubMed PMID:24201279.https://doi.org/10.1038/nature12709.
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13(4):251-262. Available from: http://www.nature.com/doifinder/10.1038/nrm3311PubMed PMID:22436748.https://doi.org/10.1038/nrm3311.
- Ghillebert R, Swinnen E, Wen J, Vandesteene L, Ramon M, Norga K, et al. The AMPK/SNF1/SnRK1 fuel gauge and energy regulator: structure, function and regulation. FEBS Journal 2011;278(21):3978-3990. Available from: http://doi.wiley.com/10.1111/j.1742-4658.2011.08315.xPubMed PMID:21883929.https://doi.org/10.1111/j.1742-4658.2011.08315.x.
- Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell 2006;124(3):471-484. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867406001085PubMed PMID:16469695.https://doi.org/10.1016/j.cell.2006.01.016.
- Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends in Biochemical Sciences 2006;31(4):215-222. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0968000406000612PubMed PMID:16540333.https://doi.org/10.1016/j.tibs.2006.02.007.
- Kadowaki T, Yamauchi T. Adiponectin Receptor Signaling: A New Layer to the Current Model. Cell Metabolism 2011;13(2):123-124. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1550413111000131PubMed PMID:21284979.https://doi.org/10.1016/j.cmet.2011.01.012.
- Goldfine AB, Kahn CR. Adiponectin: linking the fat cell to insulin sensitivity. The Lancet 2003;362(9394):1431-1432. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0140673603147277https://doi.org/10.1016/S0140-6736(03)14727-7.
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 2010;17(1):55-63. Available from: http://www.nature.com/doifinder/10.1038/nm.2277PubMed PMID:21186369.https://doi.org/10.1038/nm.2277.
- Oppert J, Nadeau A, Tremblay A, Després J, Thériault G, Dériaz O, et al. Plasma glucose, insulin, and glucagon before and after long-term overfeeding in identical twins. Metabolism 1995;44(1):96-105. Available from: http://linkinghub.elsevier.com/retrieve/pii/0026049595902953PubMed PMID:7854173.https://doi.org/10.1016/0026-0495(95)90295-3.
- Goodpaster BH, Kelley DE, Wing RR, Meier A, Thaete FL. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 1999;48(4):839-847. Available from: http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diabetes.48.4.839PubMed PMID:10102702.https://doi.org/10.2337/diabetes.48.4.839.
- Snel M, Jonker JT, Schoones J, Lamb H, Roos, A. de , Pijl H, et al. Ectopic Fat and Insulin Resistance: Pathophysiology and Effect of Diet and Lifestyle Interventions. International Journal of Endocrinology 2012;2012:1-18. Available from: http://www.hindawi.com/journals/ije/2012/983814PubMed PMID:22675355.https://doi.org/10.1155/2012/983814.
- Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proceedings of the National Academy of Sciences 2009;106(36):15430-15435. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0904944106PubMed PMID:19706383.https://doi.org/10.1073/pnas.0904944106.
- Mizuno TM, Funabashi T, Kleopoulos SP, Mobbs CV. Specific preservation of biosynthetic responses to insulin in adipose tissue may contribute to hyperleptinemia in insulin-resistant obese mice. J Nutr 2004;134(5):1045-1050.
- Kitamura T, Kahn CR, Accili D. Insulin receptor knockout mice.. Annu. Rev. Physiol 2003;65(1):313-332. Available from: http://www.annualreviews.org/doi/abs/10.1146/annurev.physiol.65.092101.142540PubMed PMID:12471165.https://doi.org/10.1146/annurev.physiol.65.092101.142540.
- McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8(3):521-533. Available from: http://www.scholaruniverse.com/ncbi-linkout?id=15331061PubMed PMID:15331061.https://doi.org/10.1016/j.cld.2004.04.004.
- Steinberg SF. Structural Basis of Protein Kinase C Isoform Function. Physiological Reviews 2008;88(4):1341-1378. Available from: http://physrev.physiology.org/cgi/doi/10.1152/physrev.00034.2007PubMed PMID:18923184.https://doi.org/10.1152/physrev.00034.2007.
- Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proceedings of the National Academy of Sciences 2011;108(39):16381-16385. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1113359108https://doi.org/10.1073/pnas.1113359108.
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006;55(Suppl 2):9-15. https://doi.org/10.2337/db06-S002.
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-associated Phosphatidylinositol 3-Kinase Activity in Muscle. Journal of Biological Chemistry 2002;277(52):50230-50236. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.M200958200PubMed PMID:12006582.https://doi.org/10.1074/jbc.M200958200.
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest 1999;103(2):253-259. Available from: http://www.jci.org/articles/view/5001https://doi.org/10.1172/JCI5001.
- Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1 H NMR spectroscopy study. Diabetologia 1999;42(1):113-116. Available from: http://link.springer.com/10.1007/s001250051123PubMed PMID:10027589.https://doi.org/10.1007/s001250051123.
- Iochida LC, Tominaga M, Matsumoto M, Sekikawa A, Sasaki H. Insulin resistance in septic rats - a study by the euglycemic clamp technique. Life Sciences 1989;45(17):1567-1573. Available from: http://linkinghub.elsevier.com/retrieve/pii/0024320589904232PubMed PMID:2685486.https://doi.org/10.1016/0024-3205(89)90423-2.
- Shangraw RE, Jahoor F, Miyoshi H, Neff WA, Stuart CA, Herndon DN, et al. Differentiation between septic and postburn insulin resistance. Metabolism 1989;38(10):983-989. Available from: http://linkinghub.elsevier.com/retrieve/pii/0026049589900103PubMed PMID:2677612.https://doi.org/10.1016/0026-0495(89)90010-3.
- Pickup JC, Crook MA. Is Type II diabetes mellitus a disease of the innate immune system. Diabetologia 1998;41(10):1241-1248. Available from: http://link.springer.com/10.1007/s001250051058PubMed PMID:9794114.https://doi.org/10.1007/s001250051058.
- Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link. Atherosclerosis 2000;148(2):209-214. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0021915099004633https://doi.org/10.1016/S0021-9150(99)00463-3.
- Hotamisligil G, Shargill N, Spiegelman B. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259(5091):87-91. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.7678183PubMed PMID:7678183.https://doi.org/10.1126/science.7678183.
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest 1995;95(5):2409-2415. Available from: http://www.jci.org/articles/view/117936https://doi.org/10.1172/JCI117936.
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307. Journal of Biological Chemistry 2000;275(12):9047-9054. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.275.12.9047PubMed PMID:10722755.https://doi.org/10.1074/jbc.275.12.9047.
- Tantiwong P, Shanmugasundaram K, Monroy A, Ghosh S, Li M, DeFronzo RA, et al. NF-κB activity in muscle from obese and type 2 diabetic subjects under basal and exercise-stimulated conditions.. Am J Physiol Endocrinol Metab 2010;299(5):794-801. Available from: http://ajpendo.physiology.org/cgi/pmidlookup?view=long&pmid=20739506PubMed PMID:20739506.https://doi.org/10.1152/ajpendo.00776.2009.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest 2003;112(12):1796-1808. Available from: http://www.jci.org/articles/view/19246PubMed PMID:14679176.https://doi.org/10.1172/JCI19246.
- Wolsk E, Mygind H, Grondahl TS, Pedersen BK, van_Hall . G IL-6 selectively stimulates fat metabolism in human skeletal muscle. Am J Physiol Endocrinol Metab 2010;299(5):832-840. https://doi.org/10.1152/ajpendo.00328.2010.
- Bézaire V, Langin D. Regulation of adipose tissue lipolysis revisited. Proc. Nutr. Soc 2009;68(04):350-360. Available from: http://www.journals.cambridge.org/abstract_S0029665109990279PubMed PMID:19698205.https://doi.org/10.1017/S0029665109990279.
- Ranjit S, Boutet E, Gandhi P, Prot M, Tamori Y, Chawla A, et al. Regulation of fat specific protein 27 by isoproterenol and TNF- to control lipolysis in murine adipocytes. The Journal of Lipid Research 2011;52(2):221-236. Available from: http://www.jlr.org/cgi/doi/10.1194/jlr.M008771https://doi.org/10.1194/jlr.M008771.
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation 2006;116(6):1494-1505. Available from: http://www.jci.org/cgi/doi/10.1172/JCI26498https://doi.org/10.1172/JCI26498.
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest 2006;116(1):115-124. Available from: http://www.jci.org/articles/view/24335PubMed PMID:16341265.https://doi.org/10.1172/JCI24335.
- Hotamisligil GS. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010;140(6):900-917. Available from: http://linkinghub.elsevier.com/retrieve/pii/S009286741000187Xhttps://doi.org/10.1016/j.cell.2010.02.034.
- Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. The Journal of Cell Biology 2010;189(5):783-794. Available from: http://www.jcb.org/cgi/doi/10.1083/jcb.201003138PubMed PMID:20513765.https://doi.org/10.1083/jcb.201003138.
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. The International Journal of Biochemistry & Cell Biology 2006;38(3):317-332. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1357272505003055PubMed PMID:16290097.https://doi.org/10.1016/j.biocel.2005.09.018.
- Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends in Cell Biology 2012;22(5):274-282. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0962892412000359PubMed PMID:22444729.https://doi.org/10.1016/j.tcb.2012.02.006.
- Ozcan U, Cao Q, Yilmaz E, Lee A, Iwakoshi NN, Ozdelen E, et al. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004;306(5695):457-461. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1103160PubMed PMID:15486293.https://doi.org/10.1126/science.1103160.
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006;313(5790):1137-1140. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1128294https://doi.org/10.1126/science.1128294.
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 2006;34(Pt 5):647-662. Available from: http://ghr.nlm.nih.gov/gene=PIK3CAPubMed PMID:17052169.https://doi.org/10.1042/BST0340647.
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010;11(1):9-22. Available from: http://www.nature.com/doifinder/10.1038/nrm2822PubMed PMID:20027184.https://doi.org/10.1038/nrm2822.
- Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell 2007;129(7):1261-1274. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867407007751PubMed PMID:17604717.https://doi.org/10.1016/j.cell.2007.06.009.
- Alessi DR, Pearce LR, García-Martínez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal 2009;2(67):27-10. Available from: http://www.scholaruniverse.com/ncbi-linkout?id=19383978PubMed PMID:19383978.https://doi.org/10.1126/scisignal.267pe27.
- Bhaskar PT, Hay N. The Two TORCs and Akt. Developmental Cell 2007;12(4):487-502. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1534580707001207PubMed PMID:17419990.https://doi.org/10.1016/j.devcel.2007.03.020.
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2010;12(1):21-35. Available from: http://www.nature.com/doifinder/10.1038/nrm3025PubMed PMID:21157483.https://doi.org/10.1038/nrm3025.
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 2012;13(6):383-396. Available from: http://www.nature.com/doifinder/10.1038/nrm3351PubMed PMID:22617471.https://doi.org/10.1038/nrm3351.
- Penkov DN, Egorov AD, Mozgovaya MN, Tkachuk VA. Insulin resistance and adipogenesis: Role of transcription and secreted factors. Biochemistry Moscow 2013;78(1):8-18. Available from: http://link.springer.com/10.1134/S0006297913010021PubMed PMID:23379555.https://doi.org/10.1134/S0006297913010021.
- Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab 2009;296(4):581-591. https://doi.org/10.1152/ajpendo.90437.2008.
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004;431(7005):200-205. Available from: http://www.nature.com/doifinder/10.1038/nature02866https://doi.org/10.1038/nature02866.
- Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends in Molecular Medicine 2007;13(6):252-259. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1471491407000664PubMed PMID:17452018.https://doi.org/10.1016/j.molmed.2007.04.002.
- Tyurin-Kuzmin PA, Morozov YI, Sukhova AA, Sagaradze GD, Zdanovskaya ND, Agaronian KM, et al. Differences in the effects of PDGF and EGF on migration and mitotic activity of NIH-3T3 fibroblasts are due to redox dependent phosphorylation of PKB/Akt, but not Erk1/2. 2014.
- Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen Peroxide Generated during Cellular Insulin Stimulation Is Integral to Activation of the Distal Insulin Signaling Cascade in 3T3-L1 Adipocytes. Journal of Biological Chemistry 2001;276(52):48662-48669. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.M105061200https://doi.org/10.1074/jbc.M105061200.
- Ravichandran LV, Esposito DL, Chen J, Quon MJ. Protein Kinase C-zeta Phosphorylates Insulin Receptor Substrate-1 and Impairs Its Ability to Activate Phosphatidylinositol 3-Kinase in Response to Insulin. Journal of Biological Chemistry 2001;276(5):3543-3549. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.M007231200https://doi.org/10.1074/jbc.M007231200.
- Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim D, et al. PKC-θ knockout mice are protected from fat-induced insulin resistance. J. Clin. Invest 2004;114(6):823-827. Available from: http://www.jci.org/articles/view/22230https://doi.org/10.1172/JCI200422230.
- Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 2011;54(6):1447-1456. Available from: http://toxnet.nlm.nih.gov/cgi-bin/sis/search/r?dbs+hsdb:@term+@rn+50-99-7PubMed PMID:21347625.https://doi.org/10.1007/s00125-011-2073-0.
- Bezy O, Tran TT, Pihlajamäki J, Suzuki R, Emanuelli B, Winnay J, et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Invest 2011;121(6):2504-2517. Available from: http://www.jci.org/articles/view/46045PubMed PMID:21576825.https://doi.org/10.1172/JCI46045.
- Frangioudakis G, Burchfield JG, Narasimhan S, Cooney GJ, Leitges M, Biden TJ, et al. Diverse roles for protein kinase C delta and protein kinase C epsilon in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C delta.. Diabetologia 2009;52(12):2616-2620. Available from: http://link.springer.com/10.1007/s00125-009-1543-0PubMed PMID:19809797.https://doi.org/10.1007/s00125-009-1543-0.
- Holland WL, Bikman BT, Wang L, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest 2011;121(5):1858-1870. Available from: http://www.jci.org/articles/view/43378https://doi.org/10.1172/JCI43378.
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest 2006;116(11):3015-3025. Available from: http://www.jci.org/cgi/doi/10.1172/JCI28898PubMed PMID:17053832.https://doi.org/10.1172/JCI28898.
- Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Molecular and Cellular Biology 2003;23(21):7794-7808. Available from: http://mcb.asm.org/cgi/doi/10.1128/MCB.23.21.7794-7808.2003PubMed PMID:14560023.https://doi.org/10.1128/MCB.23.21.7794-7808.2003.
- Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001;50(11):2563-2571. Available from: http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diabetes.50.11.2563PubMed PMID:11679435.https://doi.org/10.2337/diabetes.50.11.2563.
- Chiang S, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009;138(5):961-975. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867409007934PubMed PMID:19737522.https://doi.org/10.1016/j.cell.2009.06.046.
- Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature 2002;420(6913):333-336. Available from: http://www.nature.com/doifinder/10.1038/nature01137PubMed PMID:12447443.https://doi.org/10.1038/nature01137.
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A Stress Signaling Pathway in Adipose Tissue Regulates Hepatic Insulin Resistance. Science 2008;322(5907):1539-1543. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1160794https://doi.org/10.1126/science.1160794.
- Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proceedings of the National Academy of Sciences 2006;103(28):10741-10746. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0603509103PubMed PMID:16818881.https://doi.org/10.1073/pnas.0603509103.
- Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic Reactive Oxygen Species Regulate the Integrated Stress Response and Cell Survival. Journal of Biological Chemistry 2008;283(45):31153-31162. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.M805056200https://doi.org/10.1074/jbc.M805056200.
- Goldstein BJ, Mahadev K, Wu X. Redox Paradox: Insulin Action Is Facilitated by Insulin-Stimulated Reactive Oxygen Species With Multiple Potential Signaling Targets. Diabetes 2005;54(2):311-321. Available from: http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diabetes.54.2.311https://doi.org/10.2337/diabetes.54.2.311.
- Ткачук ВА, Тюрин-Кузьмин ПА, Белоусов ВВ, Воротников АВ. Пероксид водорода как новый вторичный посредник. Биологические мембраны: журнал мембранной и клеточной биологии. (2012); 29(1-2): 21. [Tkachuk VA, Tyurin-Kuz'min PA, Belousov VV, Vorotnikov AV. Peroksid vodoroda kak novyy vtorichnyy posrednik. Biochemistry (Moscow) Supplement. Series A, Membrane and cell biology. 2012;29(1-2):21]
Похожие статьи
Юлия Эдуардовна Азарова
Елена Юрьевна Клёсова
Александр Иванович Конопля
Юлия Эдуардовна Азарова
Елена Юрьевна Клёсова
Светлана Юрьевна Сакали
Алексей Павлович Ковалев
и другие.
Александр Юрьевич Майоров
Иван Иванович Дедов
Марина Владимировна Шестакова
Александр Сергеевич Аметов
Михаил Борисович Анциферов
и другие.
Иван Иванович Дедов
Марина Владимировна Шестакова
Гагик Радикович Галстян