This article is published under a Creative Commons license and not by the author of the article. So if you find any inaccuracies, you can correct them by updating the article.

Биоинформатические инструменты и интернет-ресурсы для оценки регуляторного потенциала полиморфных локусов, установленных полногеномными ассоциативными исследованиями мультифакториальных заболеваний (обзор)



Алексей Валерьевич Полоников
Елена Юрьевна Клёсова
Юлия Эдуардовна Азарова
Published: Jan. 1, 2021
Latest article update: Sept. 19, 2022
This article is published under the license




Abstract
Полногеномные ассоциативные исследования (genome-wide association studies, GWAS) представляют собой разновидность генетических исследований, целью которых является анализ ассоциаций между геномными вариантами и фенотипическими признаками в популяции. За последние 12 лет было установлено более 60 тысяч ассоциаций между тремя миллионами однонуклеотидных полиморфных вариантов (SNPs) и 829 заболеваниями. Тем не менее, несмотря на достигнутые успехи, большую проблему представляет вопрос патогенетической интерпретации полученных данных, поскольку абсолютное большинство локусов находятся в межгенных областях и некодирующих последовательностях генома. Цель исследования: Изучить возможности существующих биоинформатических инструментов, позволяющих оценить возможные фенотипические эффекты SNPs на определенные молекулярные функции и биологические процессы, а также имеющие патогенетическое значение для развития мультифакториальных заболеваний. Материалы и методы: Проведен анализ российской и зарубежной научной литературы по биоинформатическим методам анализа и интернет-ресурсам, необходимым для оценки регуляторного потенциала полиморфных локусов, установленных в полногеномных ассоциативных исследованиях мультифакториальных заболеваний. Результаты: В обзоре представлены основные итоги изучения спектра применения баз данных и интернет ресурсов для оценки влияния вариантов ДНК на экспрессию генов в различных тканях, метилирование ДНК, характеристики метаболомного профиля, рассмотрены алгоритмические подходы, систематизированы качественные и количественные online-инструменты, а также вычислительные методы. Заключение: Полногеномные ассоциативные исследования открыли новую эру в истории генетических исследований мультифакториальных заболеваний. Биоинформатический анализ in silico позволяет дать всестороннюю оценку эффектам SNPs и их роли в развитии того или иного фенотипического признака болезни.
Keywords
Полногеномные ассоциативные исследования, биоинформатические инструменты, ДНК-полиморфизмы, мультифакториальные заболевания
Введение.
Полногеномные ассоциативные исследования (genome-wide association studies, GWAS) представляют собой разновидность генетических исследований, целью которых является анализ ассоциаций между геномными вариантами и фенотипическими признаками в популяции [1, 2]. На сегодняшний день GWAS стали флагманом медицинской геномики открытии новых генов, которые вносят вклад в развитие полигенных мультифакториальных заболеваний. Главная цель этих исследований – формирование более глубокого понимания фундаментальных биологических основ болезни, что априори должно способствовать разработке и внедрению более эффективных способов профилактики и лечения заболеваний. Фактически при GWAS сравнивают геномы группы больных людей с геномами контрольной группы, включающей в себя аналогичных по возрасту, полу и другим признакам здоровых людей [1]. GWAS выявляют ассоциации конкретных локусов генома с признаками или заболеваниями с использованием набора однонуклеотидных полиморфизмов или SNP, максимально покрывающих геном и маркирующих блоки взаимосвязанных частых SNPs (tagSNP). Современные чипы для GWAS содержат 300000–5000000 tagSNPs с максимально возможным покрытием генома. Обнаружение ассоциации tagSNP с фенотипом означает, что один или несколько маркированных им коррелирующихся SNP должны контролировать биологические функции, которые лежат в основе выявленной ассоциации [3]. Однако трансляция накопленных в результате GWAS гено-фенотипических ассоциаций с позиции патогенеза существенно осложняется тем, что факт статистической ассоциации полиморфного варианта в геноме с каким-либо признаком не всегда является прямым свидетельством влияния гена и отражает механизмы формирования патологического процесса.
Для понимания патофизиологической взаимосвязи генетического варианта с фенотипом болезни ключевым моментом является структурно-функциональная характеристика и классификация полиморфных молекулы ДНК. Выделяют 4 основных класса полиморфных вариантов ДНК: 1) однонуклеотидные варианты (SNVs или SNPs): их число колеблется по разным данным от 3750000 до 4500000 [4]; 2) короткие (<50 нуклеотидов) инсерции и делеции (InDels): их насчитывается от 700000 до 1000000 [4]; 3) варианты числа копий (CNVs) – в основном представлены тандемными дупликациями и составляют 5-10% генома [5]; 4) структурные варианты (SVs) – составляют в среднем 13% генома [6]. Каждый вариант ДНК характеризуется собственным фенотипическим эффектом. Одни из таких вариантов хорошо изучены, другие – нет. SNPs, локализованные в области дистального промотора или 5`-нетранслируемого региона (5`-UTR), способны влиять на экспрессию гена, в котором они находятся (цис-эффекты), а также и близлежащих генов (транс-эффекты) [7]. SNPs, находящиеся в консенсусных последовательностях и регуляторных элементах (энхансерах, сайленсерах) могут влиять на сборку сплайсосомы и процесс сплайсинга [8, 9]. Варианты в 3`-нетранслируемой области (3`-UTR) и полиА-сигнальной последовательности (ААТААА) способны снижать стабильность мРНК и уменьшать ее количество, и, следовательно, и количество синтезируемого белкового продукта. Варианты, расположенные в старт- и стоп-кодонах могут существенно изменять качество трансляции, приводя к образованию более длинного или более короткого полипептида с более низкой термодинамической стабильностью по сравнению с нормальным белком [10]. Описаны необычные эффекты вариантов в межгенных элементах, способных включать или выключать экспрессию удаленных генов путем изменения статуса их метилирования.
В таблице 1 суммированы основные достижения в медицинской генетике, полученные в результате полногеномных ассоциативных исследований [1]. С помощью GWAS установлено более 10,000 надежных ассоциаций SNPs с фенотипами и болезнями. В частности, была установлена выраженная вариация показателя неравновесия по сцеплению (LD) в геноме. Выявлено, что большая доля генетической изменчивости, детерминирующей полигенные признаки и болезни, связана с влиянием частых SNPs. Был подтвержден феномен плейотропии: многие SNPs одновременно влияют на множество признаков. GWAS позволили подтвердить причинно-следственные связи и доказать ложноположительных взаимосвязи генотипов и фенотипов. С помощью GWAS было показано, что генетическая структура может имитировать географическую структуру и представлены доказательства действия естественного отбора. Сочетание GWAS с омиксными технологиями позволило установить, что 2/3 GWAS-ассоциированных SNPs расположены в гене, который не является ближайшим геном к наиболее ассоциированному SNP.
На май 2018 года каталог полногеномных ассоциативных исследований (GWAS catalog, https://www.ebi.ac.uk/gwas/home) включал более 69 млн. ассоциаций SNP с фенотипами/болезнями, обнаруженными более чем в 5000 работ и опубликованными в 3378 научных статьях. На март 2019 года база GWAS central (https://www.gwascentral.org) включала 69 986 326 ассоциаций между 2974967 уникальными SNP и 829 уникальными болезнями/фенотипами.
Несмотря на достигнутые успехи, GWAS столкнулись с очень серьезной проблемой – сложностью патофизиологической интерпретации выявленных гено-фенотипических ассоциаций. Как правило, связь между генетическим вариантом и признаком не дает непосредственной информации о гене-мишени или механизме, посредством которого данный вариант связан с фенотипом [11]. Проблема заключается в том, что наборы tagSNP, анализируемые GWAS, несмотря на максимально возможное покрытие генома, зачастую не являются причинами изучаемого заболевания. Причинный SNP может находиться где угодно в пределах гаплотипического блока, который может охватывать более 100 kb и часто содержат более 1000 отдельных SNPs [11].
Некоторые значимые ассоциации в системе полиморфизм – фенотип – метаболический путь могут быть обнаружены при рассмотрении дистантных по отношению к данному SNP генов. Так, в GWAS Catalog было зарегистрировано 12 однонуклеотидных полиморфизмов, ассоциированных с множественной миеломой [11]. Картирование этих SNP в генах, удаленных менее чем на 10 000 килобаз, не выявило генов, которые могли бы быть отнесены к какому-либо метаболическому пути. Увеличение расстояния до 400 килобаз также не дало положительных результатов. Однако, анализ SNP в связи с генами, удаленными от них более чем на 500 килобаз, привел к формированию кластеров, вовлеченных в 2 пути - «Миелома» и «Метаболизм при раке», оба имеющих близкое отношение к фенотипу. Так же, многие SNPs показали ассоциации с цветом глаз, однако, анализ связей между ними и близкорасположенными генами не дал результатов [12]. Отношение этого фенотипа к пути «меланогенез» открылось только при картировании SNPs в генах, расположенных на расстоянии 100 килобаз [13]. Парадигма «один SNP – много генов» может быть весьма полезной в обнаружении молекулярных основ фенотипического признака. Интересно, что геном существует в трехмерном измерении, и это является главным фактором, объясняющим дистанционный эффект SNPs на удаленные гены [14-16]. Накапливающиеся данные о 3D-упаковке генома позволят в ближайшем будущем определить положение SNPs в их трехмерном пространственном окружении. К сожалению, сделать это сейчас не представляется возможным в виду нехватки знаний о структурной и пространственной организации хроматина. Еще одно объяснение дистанционных эффектов SNP заключается в том, что однонуклеотидных полиморфизмы являются маркерами больших структурных вариантов, затрагивающих крупные сегменты хромосом [17].
Подавляющее большинство SNP (около 90%), ассоциированных с болезнями в результате GWAS, располагаются в некодирующих областях генома [18]. В расшифровке регуляторного потенциала некодирующих SNPs важную роль могут играть insilico инструменты, оперирующие доступными базами данных с достаточными характеристиками генной экспрессии, эпигенетических маркеров, 3D-контактов хроматина и других геномных параметров, включая гены-мишени фармакотерапии.
Использование уже разработанных биоинформатических инструментов будет способствовать пониманию функциональной значимости ассоциированных с фенотипом SNPs, что уже успешно продемонстрировано рядом работ [18-23]. Например, Watanabe K. и др. [24], опираясь на данные полногеномного исследования ассоциаций однонуклеотидных вариантов с индексом массы тела, обрабатывают результаты, используя биоинформатическую онлайн-платформу FUMA (http://fuma.ctglab.nl) для функционального аннотирования приоритетных SNPs и интерактивной визуализации взаимодействующих генов. Chen и соавторы [12] также использовали биоинформатические инструменты и ресурсы VEP, Requlome DB, ANNOVAR HaploReg для предсказания функционального и регуляторного потенциала 9 184 некодирующих вариантов базы NHGRI, и описали регулируемые механизмы 96% изученных вариантов. Более того, 3 случайно выбранных варианта из этого списка были подвержены функциональному тестированию и проявили энхансерную или сайленсерную активность. Однако фактически с начала 2015 года менее 40% GWAS использовали биоинформатические инструменты для приоритезации и предсказания функции некодирующих SNPs [25].
Биоинформатические методы и подходы к оценке регуляторного потенциала полиморфных локусов.
В обзорном исследовании Nishizaki и Boyle [26] представлена детальная характеристика современных онлайн-инструментов для оценки функциональной роли SNPs в геноме.
Функциональный анализ и интерпретация локусов, ассоциированных с болезнью в результате GWAS представляет собой сложную и многоэтапную задачу, начиная с точного картирования причинностых SNPs до исследовании функции гена-мишени. При этом на ключевых этапах данного процесса используются различные биоинформатические инструменты. На рисунке 1 представлены наиболее популярные подходы к выявлению функциональных полиморфизмов, ассоциированных с развитием патологии [3]. Данная схема анализа выявленных ассоциаций GWAS-локусов направлена на выделение приоритетных SNPs из общей массы ассоциированных с фенотипом генетических вариантов. Этот подход подразумевает интеграцию генетических данных, биоинформатического анализа и вычислительных процедур. Однако функциональная оценка некодирующих регуляторных вариантов требует пошагового применения значительного арсенала программных вычислений, в том числе обращение к большим массивам биологических данных, insilico инструментам и результатам экспериментальных молекулярных этиологий.
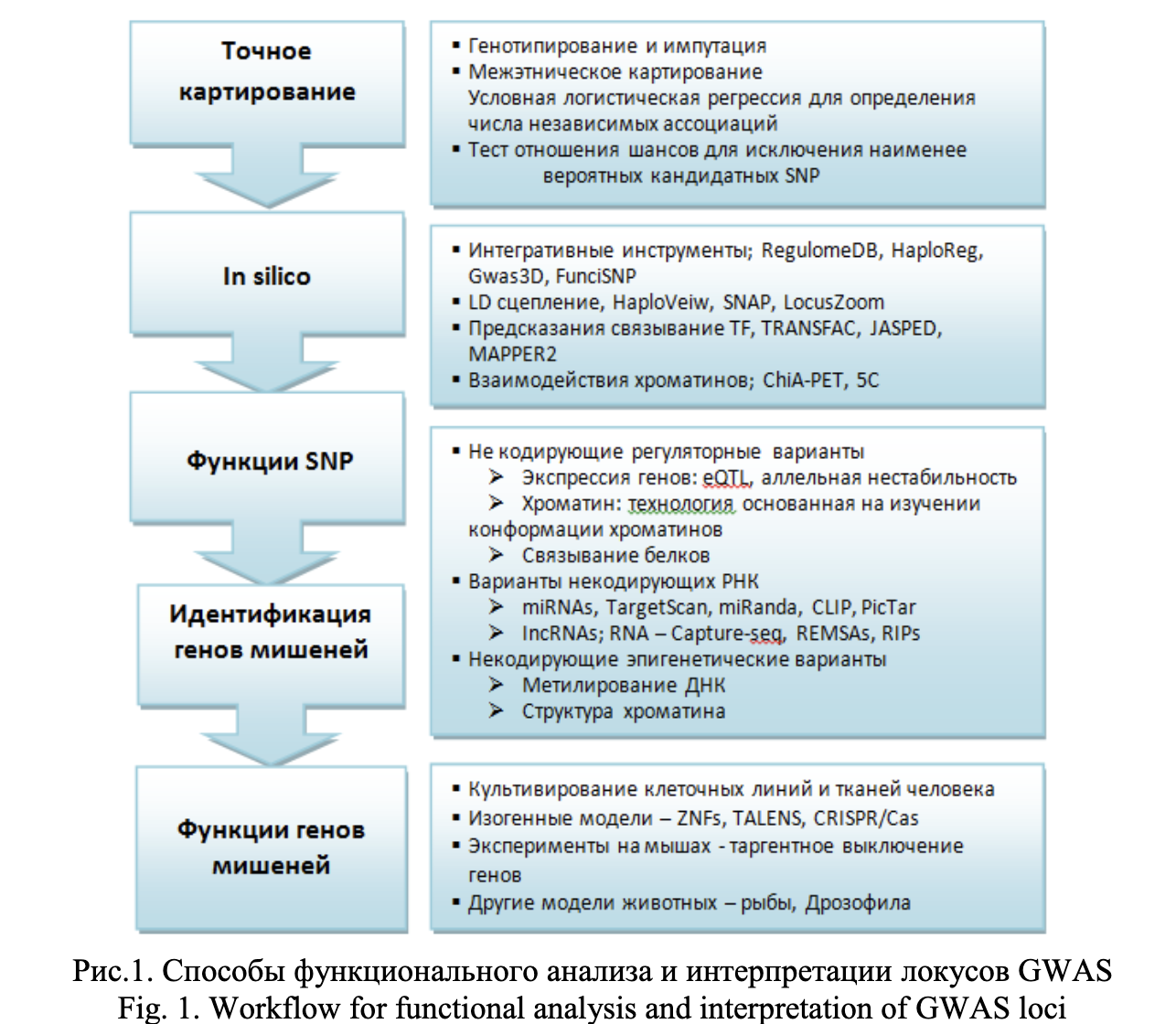
Первый этап – точное картирование, которое направлено на обнаружение т.н. причинностных SNP, влияющих на молекулярные и клеточные процессы, связанные с фенотипом болезнью. Достигается посредством, например, более “плотного” генотипирования участка генома или процедурой импутации недостающих данных генотипирования, а также статистическими процедурами (логистическая регрессия, тест отношения правдоподобия, анализ гаплотипов и др.) для выявления SNP, наиболее значимо ассоциированного с фенотипом - с наибольшим размером эффекта (effect size) и наиболее значимым уровнем значимости (P-value) влияния на фенотип [14, 27, 28].
Следующий этап – in silico аннотирование SNPs, направлен на выяснение механизма, посредством которого данный генетический вариант может влиять на экспрессию гена или активность его продукта. Сложность данной задачи заключается в интерпретации некодирующих SNP, что требует применения множества вычислительных процедур, включая анализ специальных баз данных, содержащих экспериментально подтвержденную информацию о регуляторном потенциале SNP (например, Regulome, TRANFAC, ChiA) и применение специальных биоинформатических инструментов. Сканируя мотивы в базах данных TRANSFAC, JASPAR и UniPRODE, можно легко оценить аффинность транскрипционных факторов в отношении связывания с заданными участками ДНК [19-21].
Следующий этап – оценка функциональности SNP и идентификация гена-мишени. Для некодирующих регуляторных вариантов используются: методы анализа экспрессии генов, включая выявление eQTL (локусов в геноме, ассоциированных с количественными изменениями экспрессии генов) в различных тканях с полногеномным уровнем значимости; анализ влияния 3-мерной структуры хроматина на регион ДНК с SNP, люциферазный тест, invitro тест связывания белка с ДНК [29, 30].
Для некодирующих вариантов РНК, используются инструменты для поиска их генов-мишеней и таким образом оценивают потенциал связывания микроРНК с областью SNP (TagretScan, MiRanda) [31]. Для некодирующих эпигенетических вариантов оценивают уровень метилирования ДНК, проводят иммунопреципитацию хроматина в сочетании с высокоэффективным секвенированием [32-34].
Формулировка гипотезы о биоинформатически предсказанном эффекте SNP на фенотип затем используется для тестирования этого эффекта в эксперименте. На заключительном этапе исследуют функции гена-мишени с использованием культивированных клеточных линий человеческих тканей, нокаут-моделей животных, технологии геномного редактирования (CRISPR) и других методологий [35].
Таким образом, именно некодирующие SNP являются главным и наиболее сложным объектом для анализа и биологической интерпретации связи с фенотипом болезни. Рассмотрим основные особенности регуляторного потенциала некодирующих SNP. Многие некодирующие SNPs находятся в регуляторных последовательностях генома и способны влиять на экспрессию генов на транскрипционном, посттранскрипционном и посттрансляционном уровнях [36,37]. Некодирующие варианты в энхансерах – одни из главных кандидатов для функциональной интерпретации GWAS-локусов. Регуляторные сигналы могут действовать на больших расстояниях по всему геному и вступать в контакт с промоторами-мишенями посредством трехмерной упаковки ДНК. Доступность транскрипционных факторов зависит от структурных изменений хроматина, обусловленных посттрансляционными модификациями гистонов, такими как метилирование и ацетилирование [38]. В отличие от закрытого хроматина, т.н. пермиссивный хроматин достаточно динамичен для факторов транскрипции, инициируя ремоделирование доступности специфической последовательности ДНК и обеспечивая открытую конформацию хроматина [39].
На сегодняшний день существует внушительный арсенал insilico инструментов и интернет-ресурсов для анализа регуляторного потенциала локусов, ассоциированных с болезнями [39]. Биоинформатические подходы к определению потенциальных регуляторных эффектов некодирующих SNPs были значительно усилены экспериментальными исследованиями полногеномного формата. Данные проектов ENCODE и проект Национального Института здоровья «Дорожная карта эпигенома» могут быть использованы для оценки регуляторного потенциала некодирующих вариантов и их проявления различных тканях [40,41]. Регуляторная функция полиморфизма может быть проявлением эпигенетической модификации генома, включая модификацию гистонов, регуляцию открытости хроматина, связывающую способность транскрипционных факторов. При этом можно оценить потенциальное влияние варианта посредством оценки различных геномных характеристик, таких как количественная оценка экспрессии гена в различных тканях (картирование eQTL), секвенирование хроматина (CHIP-seq технологии), секвенирование гиперчувствительных участков для ДНК-азы I, анализ взаимодействия хроматина, идентификация ДНК-мотивов, специфически связывающих транскрипционные факторы.
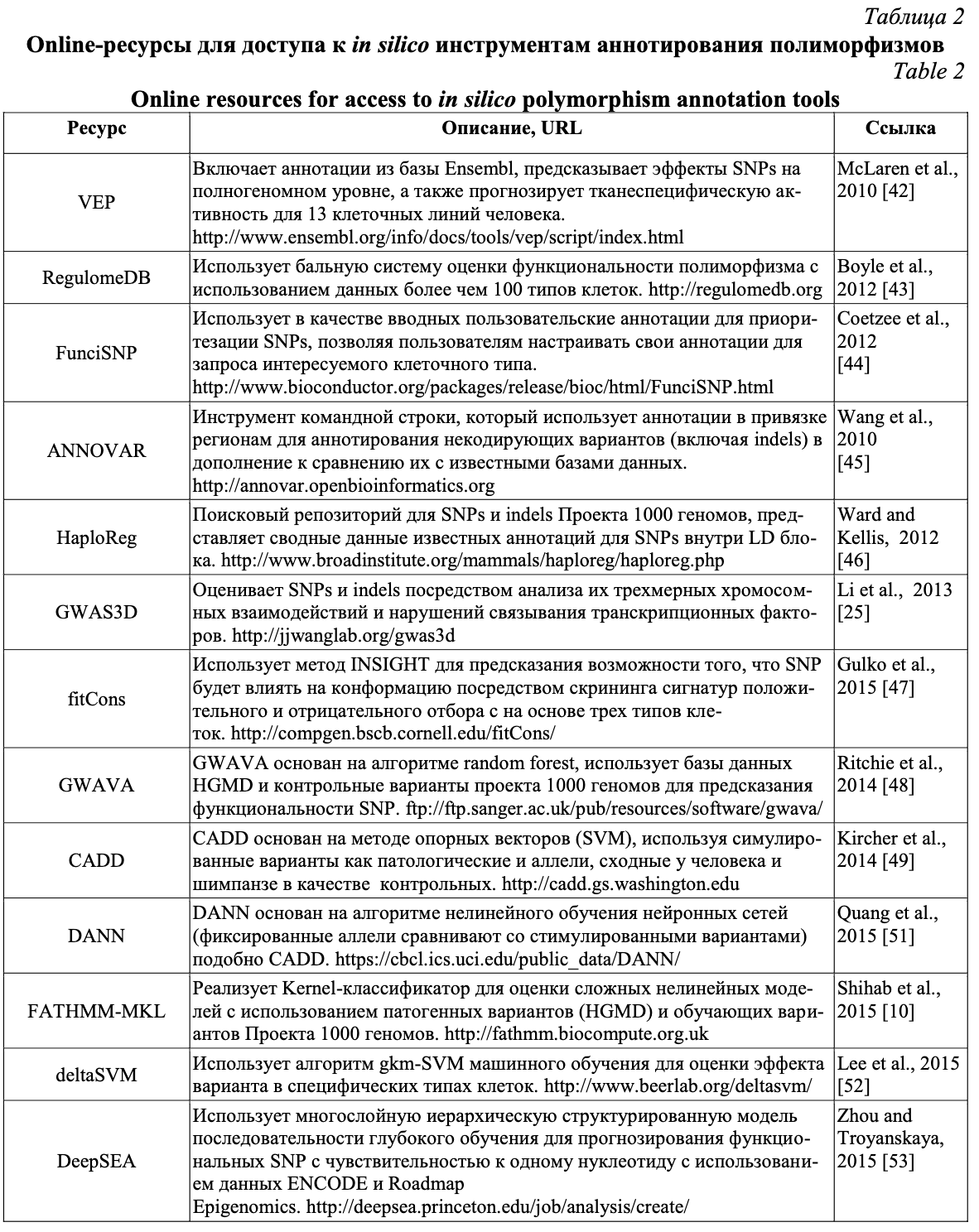
Активно использующиеся на сегодняшний день онлайн биоинформатические ресурсы направлены на оценку влияния открытого хроматина (ресурсы ENCODE, RegulomeDB, данные проекта «Дорожная карта эпигенома человека»); предсказание связывания участка транскрипционных факторов с ДНК (TRANSFAC, JASPAR); оценку ДНК-белковых взаимодействий (ENCODE, RegulomeDB, HaploReg), оценку метилирования ДНК (ENCODE, MethDB, проект «Дорожная карта эпигенома человека»); анализ экспрессии РНК, модификации гистонов и взаимодействия хроматина. В таблице 2 представлены онлайн-ресурсы для доступа к наиболее популярным биоинформатическим инструментам оценки регуляторного потенциала полиморфизмов. Данные инструменты позволяют аннотировать и предсказывать регуляторные эффекты SNPs с использованием трех основных методологических подходов: функционального аннотирования, оценки консервативности и технологии машинного обучения.
Экспериментальные подтверждения функциональности SNPs реализуется посредством современных биотехнологий. Исследования репортного гена являются дополнением к вышеописанным поисковым системам и предлагают прямое измерение функционального эффекта варианта на уровень экспрессии гена. Для этого регуляторный элемент помещают выше промотора и вводят плазмиду, содержащую интересующий ген [23]. Также, трансгенные линии животных, включая мышей и рыб, представляют собой ценный способ оценки фенотипического эффекта мутации invivo [54]. С открытием редактирования регуляторных коротких палиндромных повторов (технология геномного редактирования CRISPR), некодирующие варианты и структурные изменения могут быть легче изучены на таких более сложных модельных системах [55].
Метилирование ДНК – это фундаментальная эпигенетическая характеристика, контролирующая включение/выключение генной экспрессии. Тем не менее, взаимосвязь между характером последовательности ДНК и степенью метилирования до конца не ясна [56]. Исследования позволили выявить корреляции между профилем метилирования ДНК и индивидуальными генотипами для идентификации локусов, способных повлиять на статус метилирования генов. Было открыто множество генных локусов, объясняющих различную степень метилирования т.н. CpG-островков в зависимости от популяции или типа клеточной линии. Однако далеко не все варианты метилирования могут быть интерпретированы только лишь с учетом одних генетических факторов [56, 58]. Поэтому изучение роли SNP в формировании того или иного профиля метилирования становится одним из главных объектов для биоинформатического анализа.
Секвенирование генома нового поколения (NGS) позволяет измерить в полногеномном масштабе экспрессию генов, привязку транскрипционных факторов, доступность/открытость хроматина, модификации гистонов и метилирование ДНК. Огромные усилия были приложены для характеристики вариаций генома на транскриптомом уровне в различных клеточных линиях и тканях. Проект GENCODE (www.gencodegenes.org) содержит огромный пласт экспериментальных данных о функциональных элементах генома и представляет собой высококачественный каталог транскриптов [57]. Количественные данные о функциональной значимости вариантов могут отличаться в разных базах данных. Так, McCarthy et al [58] показали, что конкордантность Loss of Function (LoF) вариантов между ANNOVAR и VEP составляет 65%, хотя обе поисковые системы используют один и тот же набор транскриптов. Что требует стандартизации представления данных, The Sequence Ontology Progect – это первый ресурс, направленный на стандартизацию описательных характеристик генома, опирающийся на базы VEP и ANNOVAR [59].
Таким образом, вся выше представленная информация в полном объеме депонирована в сети баз данных, доступных в Internet, однако, понимание биологического смысла этой информации представляет не меньшую трудность, чем сам процесс их получения. Интерпретация выявленных ассоциаций с позиций формальной логики системы ген-мРНК-белок-метаболит в большинстве случаев крайне проблематична, поскольку абсолютное число локусов, обнаруживших ассоциации с различными фенотипами находятся в некодирующих областях генома, межгенных пространствах или в генах, не имеющих прямого отношения к изучаемому заболеванию. Путь от генотипа к фенотипу в таком случае удается проложить с помощью бионформатического инструментария, позволяющего предсказать эффект варианта ДНК на различные аспекты молекулярной жизни в микромире, включая транскрипцию, связывание транскрипционных факторов, созревание пре-мРНк, сплайсинг, трансляцию, эпигенетические модификации (метилирование ДНК, открытость хроматина). Онлайн ресурсы позволяют дать всестороннюю оценку эффектам SNPs и их роли в развитии того или иного фенотипического признака болезни.
В перспективе, увеличение объема выборок до 100000 и более человек позволит в будущих GWAS открыть новые варианты ассоциаций с известными заболеваниями, что поможет конкретизировать диагноз вплоть до его молекулярных основ и выбрать персонализированное лечение болезней. Безусловно, с течением времени, GWAS на основе SNP-панелей будут замещены GWAS на основе полногеномного секвенирования, что, вероятно, прольет свет на неизвестные по сей день аспекты взаимосвязей в системе генотип-фенотип-среда. Если 10-15 лет назад технология генотипирования была лимитирующим фактором в сфере генетических исследований, то сейчас этим фактором является полнота фенотипической характеристики обследуемых лиц. Современный биоинформатический анализ подразумевает стратификацию по фенотипическим признакам с целью выявления причинно-следственных отношений между ними и понимания того, как той или иной фактор среды опосредует воздействие генотипа на формирование признака. В конечном счете, результаты полногеномных ассоциативных исследований должны быть всесторонне проанализированы и имплементированы в практическое здравоохранение в виде более точных (по чувствительности и специфичности) диагностических предсказательных тестов и алгоритмов персонализированного лечения и профилактики социально значимой мультифакториальной патологии.
Заключение.
Полногеномные ассоциативные исследования открыли новую эру в истории генетических исследований мультифакториальных заболеваний. В данном обзоре мы представили способ поэтапного функционального анализа и интерпретации локусов GWAS, описали инструменты и online-ресурсы, позволяющие аннотировать и предсказывать регуляторные эффекты SNPs, основываясь на трех основных методиках. Выяснили, что биоинформатический анализ in silico позволяет дать всестороннюю оценку эффектам SNPs и их роли в развитии того или иного фенотипического признака болезни. Полученные результаты были успешно дополнены результатами изучения экспрессии генов в различных тканях, метилировании ДНК и характеристиками метаболомного профиля.
Информация о финансировании
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 20-15-00227).
Список литературы