This article is published under a Creative Commons license and not by the author of the article. So if you find any inaccuracies, you can correct them by updating the article.

Морфофункциональная перестройка печеночного сосудистого русла в патогенезе портальной гипертензии при циррозе печени



Д.В. Гарбузенко
Published: Jan. 1, 2014
Latest article update: Nov. 22, 2022
This article is published under the license




Abstract
Представлены современные данные о механизмах повышения печеночного сосудистого сопротивления портальному кровотоку, лежащему в основе патогенеза портальной гипертензии при циррозе печени. Показано, что, помимо грубых структурных нарушений в печени, связанных с диффузным фиброзом и формированием узлов регенерации, важную роль в этом процессе играет морфофункциональная перестройка печеночного сосудистого русла. Она характеризуется дисфункцией эндотелия и расстройством паракринного взаимодействия между активированными звездчатыми клетками печени и синусоидальными эндотелиальными клетками, ремоделированием и капилляризанией синусоидов, а также развитием внутрипеченочного ангиогенеза.
Keywords
Ангиогенез, лисфункпия энлотелия, ремоделирование синусоилов, портальная гипертензия, патогенез, иирроз печени
ГМК — гладкие мышечные клетки
ДЭ — дисфункция эндотелия
ЗКП — звездчатые клетки печени
ПГ — портальная гипертензия
СЭК — синусоидальные эндотелиальные клетки
ЦП — цирроз печени
ЭК — эндотелиальные клетки
NO — оксид азота
eNOS — эндотелиальная синтаза оксида азота
Akt — протеинкиназа В
PAF — фактор активации тромбоцитов
PDGF — тромбоцитарный фактор роста
HGF — гепатоцитарный фактор роста VEGF — фактор роста сосудистого эндотелия IGF — инсулиноподобный фактор роста TGF — трансформирующий фактор роста EGF — эпидермальный фактор роста FGF — фактор роста фибробластов
CTGF — фактор роста соединительной ткани HIF — фактор, индуцированный гипоксией ROS — реактивные формы кислорода PIGF — плацентарный фактор роста INF — фактор некроза опухоли
Портальная гипертензия (ПГ) может встречаться при различных заболеваниях, сопровождающихся повышением давления в системе воротной вены. ПГ характеризуется рядом тяжелых осложнений, среди которых наиболее частым и опасным является кровотечение из варикозно-расширенных вен пищевода. При циррозе печени (ЦП) в основе присущих ей гемодинамических нарушений лежит повышение печеночного сосудистого сопротивления портальному кровотоку. Последующее формирование естественных портосистемных шунтов и развитие гипердинамического циркуляторного статуса является следствием сложных процессов ангиогенеза, ремоделирования сосудов и дисфункции эндотелия (ДЭ), способствуя дальнейшему прогрессированию ПГ [1].
Считается, что главным местом сопротивления портальному кровотоку при ЦП служат патологически-измененные синусоиды. Синусоидальные эндотелиальные клетки (СЭК) активируются и приобретают сосудосуживающий фенотип. В этой ситуации чувствительность СЭК к эндогенным вазоконстрикторам, в частности эндотелину, норадреналину, ангиотензину II, вазопрессину, лейкотриенам, тромбоксану А, повышается, а производство ими оксида азота (NO) — наиболее изученного вазодилататора, участвующего в регуляции тонуса печеночных сосудов, наоборот, снижается [2]. Причина может заключаться в недостаточной активности эндотелиальной синтазы NO (eNOS) из-за повышенного взаимодействия ее молекулы с кавеолином-1. Кроме того, стимуляция эндотелином-1 киназ рецепторов, сопряженных с G-белками 2-го класса, приводит к ингибированию фосфорилирования протеинкиназы В (Akt) и снижению продукции NO.
Одним из главных факторов ДЭ синусоидов при ЦП является внутрипеченочный оксидантный стресс, способствующий уменьшению экспрессии eNOS и биодоступности NO. Например, циклооксигеназа, участвуя в синтезе тромбоксана Aj, как и избыточная стимуляция Rho-киназы, ингибирует фосфорилирование Akt в эндотелиальных клетках (ЭК), существенно подавляя Akt-eNOS-сигнализацию. Асимметричный диметиларгинин, угнетая активность eNOS, вызывает генерацию пероксинитрита, а пониженная экспрессия тетрагидробиоптерина способствует продукции eNOS вместо NO кислорода. Кроме того, сообщалось, что возможной причиной недостаточной биодоступности NO могут быть редукция активности «фермента, сберегающего NO» супероксиддисмутазы и повышение в сыворотки крови уровня гомоцистеина вследствие уменьшения экспрессии ферментов цистатионин-у-лиазы и цистатионин-З-синтазы [3].
Важная роль в синусоидальной микроциркуляции при ЦП придается активированным звездчатым клеткам печени (ЗКП) и их паракринному взаимодействию с СЭК. В данной патологической ситуации нарушение структуры и функции ЗКП сопровождается потерей накоплений ретиноидов и трансформацией ЗКП в миофибробласты. Активированные ЗКП начинают выполнять роль перицитов, что подтверждается экспрессией ими таких фенотипических маркеров этого типа клеток, как п-гладкомышечный актин, десмин, NG2, глиальный фибриллярный кислый протеин, появлением или увеличением на их поверхности числа рецепторов для факторов роста, цитокинов и эндотелина, а также ряда молекул адгезии клеток [4].
Располагаясь в субэндотелиальном пространстве Диссе между СЭК и гепатоцитами, ЗКП благодаря своим длинным ветвящимся цитоплазматическим отросткам, простирающимся вдоль и вокруг синусоидов, контактируют с нервными окончаниями, содержащими такие нейропептиды, как вещество Р, вазоактивный интестинальный пептид, соматостатин, холецистокинин, нейротензин, NO, кальцитонин ген-связанный пептид и нейропептид Y. Некоторые вазоактивные вещества способны регулировать тонус ЗКП. Причем, если эндотелии-1, вещество Р, ангиотензин II, норадреналин, простагландин F2, тромбоксан Aj, фактор активации тромбоцитов (PAF), тромбин вызывают констрикцию ЗКП, то NO, ацетилхолин, вазоактивный интестинальный пептид, монооксид углерода, сероводород, простагландин Е2 и адреномедуллин содействуют расслаблению этих клеток [5].
В сокращении ЗКП принимает участие миозин II типа, а сам процесс регулируется как зависимыми, так и независимыми от кальция механизмами. При зависимом от кальция пути фосфорилирование легких цепей миозина происходит с помощью киназы легких цепей миозина, активированной в ответ на увеличение концентрации внутриклеточного кальция ([Са2+]і) и последующего формирования кальций/кальмодулинового комплекса. В то же время два независимых от кальция пути связаны с ингибированием активности фосфатазы легких цепей миозина посредством активации Rho-киназы и протеинкиназы С [6].
К модуляторам тонуса ЗКП относятся прежде всего мощные вазоконстрикторы — эндотелины. Это семейство 3 гомологичных олигопептидов, являющихся продуктами протеолиза их предшественника «большого эндотелина» под влиянием эндоте- линпревращающего фермента. Они действуют через рецепторы 2 типов (А и В), сопряженных с белком G, которые хорошо выражены в ЗКП. Наиболее изученным является эндотелии-1, основным местом синтеза которого при ЦП служат чувствительные к нему активированные ЗКП. Стимуляция рецепторов эндотелина А приводит к их сокращению и пролиферации [7]. Аналогичное действие оказывает и ангиотензин II. Его синтез ЗКП при ЦП увеличивается в результате повышенной экспрессии ангиотен- зинпревращающего фермента [8]. Констрикция ЗКП также может быть обусловлена уменьшением продукции и/или биодоступности NO в цирротически измененной печени. Напротив, гиперпродукция монооксида углерода клетками Купфера в результате паракринного влияния как на ЗКП, так и СЭК вызывает расширение синусоидов и, таким образом, уменьшение печеночного сосудистого сопротивления [9].
Повышенная подвижность и миграция ЗКП при ЦП приводят к увеличению плотности покрытия ими синусоидов, способствуя их ремоделированию [10]. Важную роль в этом процессе играет изменение структуры мембраны ЗКП. Пространственно управляемая полимеризация актина лежит в основе подвижности клеток и отвечает за формирование клеточных протрузий — ла- меллиподий, а также филлоподий, образованных радиально ориентированными пучками актиновых филаментов, встроенных в ламеллиподии. Семейство Rho гуанозинтрифосфатаз, включающее RhoA (Rho), Rael (Rae) и Cdc42, регулирует формирование этих структур. Показано, что если Rac способствует миграции ЗКП за счет образования филлоподий, то Rho вызывает устойчивость к ингибирующему действию NO, а также восстанавливает хемотаксический ответ на тромбоцитарный фактор роста (PDGF) в отсутствие функциональной Rac [11].
Ключевую роль в пролиферации, миграции, подвижности и рекрутинге ЗКП играет PDGF. Он секретируется СЭК и связывается со своим родственным рецептором (PDGFR-ß), расположенном на перицитах, в частности за счет сигнального пути эфрин-В/Ер11В4 [12]. Причем, если стимуляция киназы Rak-1, киназы МЕК и внеклеточных регулируемых сигналом киназ ERK, вызванная активацией PDGFR-ß, приводит к пролиферации ЗКП, то стимуляция фосфатидилинозитол-3-киназы — к хемотаксису [13]. Кроме того, показано, что трансмембранный рецептор нейропилин-1 также способствует хемотаксическому ответу на PDGF [14].
Активированные ЗКП служат богатым источником полипептидов, эйкозаноидов, а также различных других малых молекул с паракринной, юкстакринной, аутокринной функцией или хемоаттрактантной активностью, к которым относятся следующие:
- полипептиды, которые усиливают пролиферацию аутокринным и паракринным способом: гепатоцитарный фактор роста (HGF), фактор роста сосудистого эндотелия (VEGF), эндотелии-1, инсулиноподобный фактор роста (IGF)-II, и, возможно, трансформирующий фактор роста (TGF)-a, эпидермальный фактор роста (EGF) и кислотный фактор роста фибробластов (aFGF);
- члены суперсемейства TGF-ß;
- нейротрофины;
- гемопоэтические факторы роста, такие как эритропоэтин [15].
При повреждении печени активированные ЗКП пролиферируют и мигрируют в зоны воспаления и некрозов гепатоцитов, продуцируя избыточное количество компонентов внеклеточного матрикса. Главными веществами, регулирующими этот процесс, являются TGF-ßl, PDGF, а также фактор роста соединительной ткани (CTGF) и FGF [16].
В целом существует 3 основных источника фиброгенных клеток в печени: 1) эндогенные (резидентные) фибробласты или миофибробластоподобные клетки, в основном представленные активированными ЗКП и портальными фибробластами; 2) клетки, полученные в результате так называемого эпителиально-мезенхимального транзита, который может приводить к трансдифференцировке паренхиматозных клеток; 3) гемопоэтические и мезенхимальные стволовые клетки костного мозга [17].
В 1983 г. А. Rappaport и соавт. [18] одними из первых описали коллатеральную микроциркуляцию в цирротически-измененной печени. В настоящее время патологический ангиогенез хорошо охарактеризован как при экспериментальном фиброзе печени [19], так и в клинике у больных хроническими вирусными и аутоиммунными заболеваниями печени [20], неалкогольным стеатогепатитом [21].
Ангиогенез — сложный физиологический процесс образования новых кровеносных сосудов из ранее существующих. Он осуществляется посредством активации ЭК, экспрессии в них протеаз, разрушения внеклеточного матрикса, пролиферации, миграции и образования ЭК первичных высокопроницаемых сосудистых структур, которые после стабилизации и «взросления» за счет привлечения перицитов и гладких мышечных клеток (ГМК), трансформируются в трехмерную сосудистую сеть [22].
Основным индуктором ангиогенеза, как в физиологических условиях, так и при различных патологических состояниях, является гипоксия. Клетки реагируют на недостаток кислорода несколькими механизмами, в том числе накоплением факторов, индуцированных гипоксией (HIFs), что стимулирует экспрессию ангиогенных факторов роста. Семейство HIFs включает 3 а-субъединицы, которые сопряжены с общей ß-субъединицей (HIF-lß). Если HIF-Іа выражен повсеместно, то HIF-2a обнаружен в ограниченном типе клеток, в частности в сосудистых ЭК, гепатоцитах, пневмоцитах II типа и макрофагах. Роль HIF-За в гипоксических состояниях изучена плохо [23].
NADPH-оксидаза является важным медиатором ангиогенной сигнализации. Отмечалось, что ее повышенная экспрессия в результате фосфорилирования цитозольного компонента p47phox приводит к увеличению формирования реактивных форм кислорода (ROS), что способствует индукции HIF-la, активации рецепторов VEGF (VEGFR) и трансактивации рецепторов EGF [24].
Недавно показана важная роль микроРНК в регуляции ответа клеток на гипоксию. В частности, Let-7 и mlR-103/107 посредством нацеливания белка argonaute 1 способствуют индукции VEGF [25].
К наиболее исследованным ангиогенным факторам роста относится семейство VEGF, состоящее из 5 гомологов: VEGF-A, В, С, D и плацентарного фактора роста (PIGF). VEGF стимулирует как физиологический, так и патологический ангиогенез. Все представители этого семейства соединены с различными родственными им рецепторами: VEGFR-1 (Flt-1), VEGFR-2 (KOR/ Flk-1), VEGFR-3 (Flt-4), из которых только первые 2 отвечают за передачу ангиогенных сигналов. При этом связывание VEGF-А с VEGFR-2 и повышение проницаемости сосудов посредством NO являются механизмами, запускающими процессы ангио- и ва- скулогенеза.
В отличие от VEGF PIGF соединенный с VEGFR-1 индуцирует исключительно патологический ангиогенез, прямо или опосредованно влияя на многочисленные типы клеток, в том числе ЭК. Кроме того, предполагается, что, нарушая связь VEGF с VEGFR-1, PIGF делает VEGF более доступным к соединению с VEGFR-2. Поскольку как PIGF, так и VEGF индуцируют фосфорилирование тирозиновых остатков VEGFR-1, можно предположить, что именно через него они передают определенные ангиогенные сигналы.
Разные механизмы лежат в основе синергизма между PIGF и VEGF. PIGF активирует VEGFR-1 и индуцирует межмолекулярное взаимодействие VEGFR-1 и VEGFR-2, укрепляя связь последнего с VEGF. PIGF как субъединица PIGF/VEGF гетеродимеров вызывает образование VEGFR-1/2 гетеродимеров, которые трансфосфорилируют друг друга в ходе интрамолекулярной реакции. Кроме того, продуцируя PIGF, ЭК способствует увеличению своей чувствительности к VEGF. Помимо ЭК секретировать PIGF способны и соседние стромальные или воспалительные клетки.
PIGF может непосредственно воздействовать на ГМК, экспрессирующие VEGFR-1, и косвенно влиять на их пролиферацию и миграцию через высвобождение цитокинов активированными ЭК. Это приводит к скоплению ГМК вокруг образующихся сосудов, делая их зрелыми, прочными и герметичными.
PIGF также мобилизует несущие VEGFR-1 гемопоэтические прогениторные клетки из костного мозга и опосредованно, через повышенную экспрессию VEGF, привлекает в ишемические ткани эндотелиальные прогениторные клетки, несущие VEGFR-2. Кроме того, PIGF является хемоаттрактантом для моноцитов и макрофагов, которые экспрессируют VEGFR-1 [26].
Стимулировать ангиогенез также способны члены семейства FGF. Клеточный ответ на FGFs происходит через специфическое связывание с рецепторами FGF (FGFR), обладающими внутренней тирозинкиназной активностью. Димеризация FGFR служит предпосылкой для фосфорилирования и активации сигнальных молекул при участии связывающих гепарин белков. Это вызывает миграцию, пролиферацию, дифференцирование клеток и разрушение внеклеточного матрикса. Следует отметить, что если члены семейства VEGF задействованы, главным образом в формировании капилляров, то FGFs вовлечены прежде всего в артериогенез [27].
Хотя ангиогенный эффект PDGF не столь выражен, как у VEGF, PIGF и FGF, исследования in vivo показали, что он может стимулировать формирование кровеносных сосудов и регулировать их тонус [28].
В координации ангиогенных процессов важную роль играют экспрессируемый ЭК тирозинкиназный рецептор Тіе2 (Тек) и его лиганды ангиопоэтины. Ангиопоэтин 1-го типа индуцирует миграцию и ингибирует апоптоз ЭК, а также стимулирует формирование их ростка, способствуя стабилизации сосудов. При этом в обусловленную им активацию Akt и активируемых митогеном прогеинкиназ (р42/р44 МАРК, или ERK2 и ERK1) и последующую модуляцию миграции ЭК и ангиогенез непосредственно вовлечена NADPH-оксидаза [29]. Напротив, ангиопоэтин 2-го типа посредством смещения ЭК от устойчивого состояния до пролиферативного фенотипа вызывает дестабилизацию сосудов. Вместе с тем при наличии VEGF он может также стимулировать ангиогенез [30].
Интегрины aVß3 и aVß5 — рецепторы адгезии клеток являются положительными регуляторами ангиогенеза. Они принимают участие в миграции и пролиферации ЭК и образовании новых кровеносных сосудов [31].
Белок клеточной адгезии эндотелия сосудов ѴЕ-кадгерин способствует межклеточному контакту во время неоваскуляризации и управляет проходом молекул через эндотелиальную выстилку [32].
Тромбоспондин-1 — один из 5 известных тромбоспондинов — адгезивных белков, регулирующих взаимодействие клеток между собой и с внеклеточным матриксом. При прогрессировании ЦП экспрессия тромбоспондина-1 усиливается, соответствуя тяжести фиброза и сильно коррелируя с выраженностью ангиогенеза. Вместе с тем точная роль тромбоспондина-1 в этом процессе не определена. Он может функционировать как ингибитор и промотор ангиогенеза, что зависит от его концентрации, а также от типа и числа рецепторов, представленных на поверхности ЭК [33].
Ангиостатин — продукт деградации плазминогена, а эндостатин — С-концевой фрагмент коллагена XVIII типа ингибируют индуцированную VEGF и FGF миграцию циркулирующих ЭК без вмешательства в ключевой внутриклеточный сигнальный каскад, вовлеченный в миграцию и пролиферацию клеток [34].
Толлподобный рецептор 4, служащий для распознавания бактериального липополисахарида, экспрессируется СЭК и при ЦП вовлечен в ассоциированный с фиброзом ангиогенез. Эти свойства он проявляет через родственный цитозольный адаптерный белок MyD88, который участвует в продукции внеклеточной протеазы, регулирующей инвазивную способность СЭК [35].
Печеночная алелиновая система (апелин/АРІ-рецептор) — связующее звено между хроническим воспалением и последующими фиброгенными и ангиогенными процессами, происходящими при ЦП. С одной стороны, гипоксия и воспаление инициируют печеночную экспрессию API, с другой — его активация опосредует индукцию профиброгенных генов, пролиферацию ЗКП и секрецию проангиогенных факторов [36].
Аквапорин-1 — представитель семейства интегральных мембранных белков, формирующих поры в мембранах клеток, избыточно экспрессируется в цирротически-измененной печени и стимулирует ангиогенез путем повышения эндотелиальной инвазии [37].
Известно, что хемокины подсемейства СХС принимают участие в ангиогенезе. Причем, если ЕЬК[*]-положительные хемокины его стимулируют, то ELR-отрицательные — подавляют [38].
Нейропилины (1 и 2) представляют собой трансмембранные рецепторы с большими внеклеточными доменами, которые взаимодействуют с секретируемыми семафоринами 3-го класса, а также с VEGF и его рецепторами VEGFR-1 и VEGFR-2. В сосудистом русле нейропилин-1 экспрессируется главным образом в
артериальном эндотелии, тогда как нейропилин-2 — в венозном и лимфатическом. Несмотря на то что нейропилины хорошо выражены в местах как физиологического, так и патологического ангиогенеза, их роль в этом процессе до конца не изучена [39].
Проявления патологического ангиогенеза при хронических заболеваниях печени существенно отличаются от соответствующих процессов в других органах и тканях, что объясняется рядом причин: 1) уникальным фенотипическим профилем и функциональной ролью активированных ЗКП и других печеночных миофибробластов; 2) наличием двух различных капиллярных структур, а именно, синусоидов, имеющих фенестрированный, лишенный базальной мембраны эндотелий, и сосудов, выстланных непрерывным эндотелием; 3) существованием подобного ангио- поэтину белка 3 (ANGPTL3), специфичного для печени ангиогенного фактора.
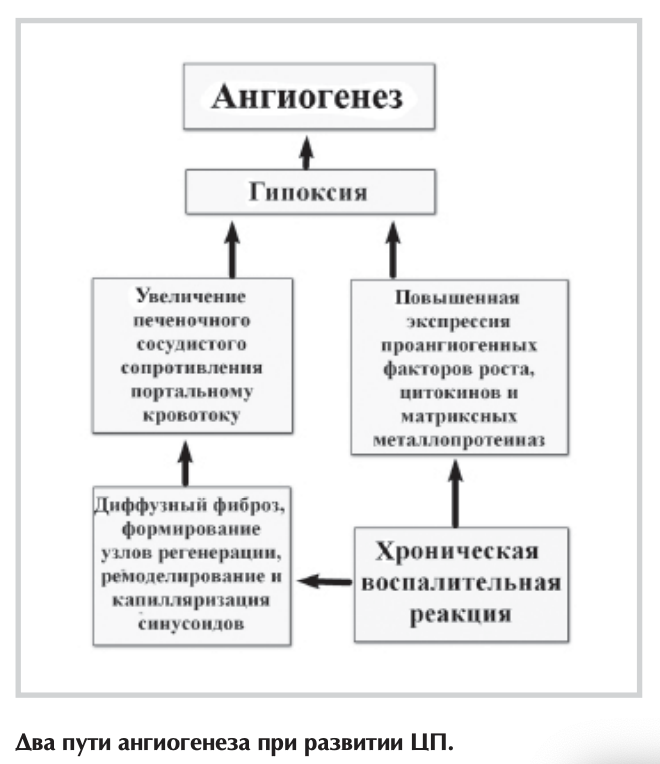
При этом выделяют 2 основных пути формирования новых кровеносных сосудов во время развития ЦП (см. рисунок) [40].
Один из них связан с повышенной экспрессией проангио- генных факторов роста, цитокинов и матриксных металлопротеиназ на фоне хронического воспаления. Провоспалительные медиаторы, вырабатываемые клетками Купфера, тучными клетками, а также лейкоцитами, могут проявлять ангиогенный ответ за счет индукции и увеличенной транскрипционной активности HIF-la [41].
Считается, что в обычном состоянии макрофаги непосредственного участия в ангиогенезе не принимают. Напротив, при ЦП активированные клетки Купфера способствуют образованию новых кровеносных сосудов через выработку ими цитокинов, ROS и PAF [42]. Причем секретируемый ими «-фактор некроза опухоли (TNF-a) через путь MAPK/ERK вызывает миграцию клеток, а также регулирует апоптоз и ангиогенез [43]. Увеличение содержания ROS в печени стимулирует ангиогенез за счет повышенной экспрессии TNF-a, NO, HIF-1 и VEGF [44]. PAF, активируя ядерный фактор транскрипции NF-xB, содействует выработке VEGF [45]. Тучные клетки участвуют в образовании новых кровеносных сосудов посредством продукции ими гепарина, гистамина, триптазы, цитокинов (TGF-ßl, TNF-a, интерлейкинов) и VEGF. Описана также их способность увеличивать число СЭК in vitro [46]. При хроническом воспалении печени отмечена повышенная экспрессия хемокинов, которая регулируется провоспалительными цитокинами, факторами роста, протеазами и прод уктами оксидантного стресса. Благодаря этому лейкоциты за счет экстравазации могут проникать в ткань печени, где продуцируют такие ангиогенные факторы, как VEGF, FIGF, PDGF, FGF, TGF-ßl, EGF, ангиопоэтин 2-го типа и различные интерлейкины [47].
С одной стороны, гипоксия, вызванная стимуляцией HIF- la, активирует ЗКП, что приводит к выработке различных ангиогенных и фиброгенных факторов (PIGF, VEGF, NO, HGF, PDGF) [48], содействуя развитию как ангиогенеза, так и прогрессированию фиброза печени [49]. С другой стороны, диффузный фиброз, формирование узлов регенерации, а также калилляриза- ция синусоидов вызывают увеличение печеночного сосудистого сопротивления и ухудшают доставку кислорода клеткам печени [50]. Накопление HIFs, в частности HIF-la, повышает экспрессию VEGF и ангиопоэтина 1-го типа и их родственных рецепторов на активированных ЗКП. Это приводит к привлечению и стимуляции СЭК, что стабилизирует новообразованные сосуды и обеспечивает их прочность [51]. В свою очередь СЭК вырабатывают PDGF и TGF-ß, способствуя привлечению и миграции ЗКП — процессу, который включает опосредованную ROS активацию внеклеточных регулируемых сигналом киназ ERK и с-Jun- NH2-repMu калькой протеинкиназы (INK) с последующим зависимым от HIF-la синтезом VEGF [52].
Соответственно выделяют 2 патоморфологические фазы ангиогенного процесса при развитии ЦП. Первоначально образование сосудов происходит в формирующихся неполных септах, где сопутствующая экспрессия VEGF, Flk-1 и Tie-2 ограничена активированными ЗКП. На более позднем этапе ангиогенез встречается в больших мостовидных септах, а проявления этой проангиогенной панели определяются в СЭК и направлены на стабилизацию новообразованных сосудов [53]. Причем одни из них располагаются вокруг и внутри фиброзных септ и, вероятно, необходимы для компенсации недостаточного кровотока в печени. Другие, формирующие внутрипеченочные портокавальные шунты, несут кровь в обход синусоидов. Из-за снижения доставки кислорода и питательных веществ к тканям печени и ограничения свободного обмена между гепатоцитами и синусоидами они могут привести к ее дисфункции, несмотря на свою декомпрессивную роль [54]. В последние годы установлено, что ЭК-предшественники, произведенные стволовыми клетками костного мозга, способны вызывать in situ неоваскуляризацию как в физиологических, так и патологических условиях (постнатальный васкулогенез). В частности, они могут усиливать ангиогенез у больных ЦП, стимулируя СЭК посредством секреции факторов с паракринной функцией, таких как PDGF и VEGF [55]. Однако их ангиогенная способность у пациентов данной категории значительно снижена, особенно при тяжелых нарушениях функции печени. Возможно, это связано с тем, что хроническое воспаление стимулирует выпуск ангиогенных факторов резидентными ЗКП и СЭК, и подавляет мобилизацию ЭК-предшественников костного мозга в кровоток [56]. Таким образом, помимо грубых структурных изменений в цирротически-измененной печени, связанных с диффузным фиброзом и формированием узлов регенерации, важную роль в повышении печеночного сосудистого сопротивления портальному кровотоку играет ДЭ и нарушение паракринного взаимодействия между активированными ЗКП и СЭК, а также ремоделирование и капилляризация синусоидов. При этом развитие внутрипеченочного ангиогенеза можно рассматривать как компенсаторный механизм, направленный на декомпрессию портальной системы. Вместе с тем новообразованные сосуды, несущие кровь в обход синусоидов, не способны обеспечить кислородом и питательными веществами ткани печени, что приводит к прогрессированию заболевания. Всесторонняя оценка морфофункциональных изменений печеночного русла при формировании ЦП позволит разработать новые методы коррекции характерных для него гемодинамических нарушений, в частности, повысить эффективность лечебных мероприятий, направленных на профилактику осложнений портальной гипертензии.
ЛИТЕРАТУРА
- Гарбузенко Д.В. Механизмы адаптации сосудистого русла к гемодинамическим нарушениям при портальной гипертензии. Вести РАМН 2013; 1: 52-57.
- Garcia-Pagan J. С., Gracia-Sancho J., Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol 2012; 57 (2): 458-461.
- Hu L.S., George J., Wang J.H. Current concepts on the role of nitric oxide in portal hypertension. World J Gastroenterol 2013; 19(11): 1707-1717.
- Hellerbrand C. Hepatic stellate cells-the pericytes in the liver. Pflügers Arch 2013; 465 (6): 775-778.
- Ueno T, Bioulac-Sage P., Balabaud C., Rosenbaum J. Innervation of the sinusoidal wall: regulation of the sinusoidal diameter. Anat Rec A Discov Mol Cell Evol Biol 2004; 280 (1): 868-873.
- lizuka M., Murata T, Hori M., Ozaki H. Increased contractility of hepatic stellate cells in cirrhosis is mediated by enhanced Ca2+- dependent and Ca2+-sensitization pathways. Am J Physiol Gastrointest Liver Physiol 2011; 300 (6): 1010—1021.
- Takashimizu S., Kojima S, Nishizaki Y. et al. Effect of endothelin A receptor antagonist on hepatic hemodynamics in cirrhotic rats. Impheations for endothelin-1 in portal hypertension. Tokai J Exp Clin Med 2011; 36 (2): 37-43.
- Lugo-Baruqui A., Munoz-Valle J.F., Arevalo-Gallegos S., Armenddriz-Borunda J. Role of angiotensin II in liver fibrosis- induced portal hypertension and therapeutic implications. Hepatol Res 2010; 40 (1): 95-104.
- Reynaert H, Urbain D., Geerts A. Regulation of sinusoidal perfusion in portal hypertension. Anat Rec 2008; 291 (6): 693—698.
- Lee J.S., Semela D., Iredale J., Shah V.H. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology 2007; 45 (3): 817-825.
- Lee J.S., Decker N.K., Chatterjee S. et al. Mechanismus of nitric oxide interplay with Rho GTFase tamely members in modulation of actinmembrane dynamics in pericytes and fibroblasts. Am J Pathol 2005; 166 (6): 1861-1870.
- Semela D., Das A., Langer D. et al. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology 2008; 135 (2): 671—679.
- PDGF and signal transduction in hepatic stellate cells. Front Biosci 2002; 7:1720—1726.
- Cao S., Yaqoob U, Das A. etal. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest 2010; 120 (7): 2379-2394.
- Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the fiver. Physiol Rev 2008; 88 (1): 125—172.
- Яковенко Э.П., Яковенко A.B., Иванов A.H. и др. Фиброз печени: механизмы развития и вопросы терапии. Фарматека 2011; 225 (12): 16-22.
- Svegliati-Baroni G., De Minicis S., Marzioni M. Hepatic fibrogenesis in response to chronic fiver injury: novel insights on the role of cell-to-cell interaction and transition. Liver Int 2008; 28 (8): 1052-1064.
- Rappaport A.M., MacPhee P.J., Fisher M.M., Phillips M.J. The scarring of the fiver acini (Cirrhosis). Tridimensional and microcirculatory considerations. Virchows Arch A Pathol Anat Histopathol 1983; 402 (2): 107-137.
- Lemos Q. T, Andrade Z.A. Angiogenesis and experimental hepatic fibrosis. Inst. Oswaldo Cruz 2010; 105 (5): 611—614.
- Medina J., Arroyo A.G., Sanchez-Madrid E, Moreno-Otero R. Angiogenesis in chronic inflammatory fiver disease. Hepatology 2004; 39 (5): 1185-1195.
- Ciupinska-Kajor M., Hartleb M., Kajor M. et al. Hepatic angiogenesis and fibrosis are common features in morbidly obese patients. Hepatol Int 2013; 7 (1): 233—240.
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 2007; 6 (4): 273—286.
- Skuli N., Majmundar A.J., Krock B.L. et al. Endothelial HIF-2a regulates murine pathological angiogenesis and revascularization processes. J Clin Invest 2012; 122 (4): 1427—1443.
- Brandes R.P., Miller F.J., Beer S. et al. The vascular NADPH oxidase subunit p47phox is involved in redox-mediated gene expression. Free Radio Biol Med 2002; 32 (11): 1116—1122.
- Chen Z., Lai T.C., Jan Y.H et al. Hypoxia-responsive miRNAs target argonaute 1 to promote angiogenesis. J Clin Invest 2013; 123 (3): 1057-167.
- Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med 2004; 255 (5): 538-561.
- Klein S., Roghani M., Rifkin D.B. Fibroblast growth factors as angiogenesis factors: new insights into their mechanism of action. EXS 1997; 79:159-192.
- Hellberg C, OstmanA., Heldin C.H. PDGF and vessel maturation. Recent Results Cancer Res 2010; 180:103—114.
- Chen J.X., Zeng H, Lawrence M.L. et al. Angiopoietin-1 -induced angiogenesis is modulated by endothelial NADPH oxidase. Am J Physiol Heart Circ Physiol 2006; 291 (4): 1563-1572.
- Ramsauer M., D’Amore P.A. Contextual role for angiopoietins and TGFßl in blood vessel stabilization. J Cell Sei 2007; 120 (Pt 10): 1810-1817.
- Patsenker E., Popov Y, Stickel F. et al. Pharmacological inhibition of integrin avß3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology 2009; 50 (5): 1501—1511.
- Kevil C.G., Payne D.K., Mire E., Alexander J.S. Vascular permeability factor/vascular endothelial cell growth factor- mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 1998; 273 (24): 15099-15103.
- Elpek G.O., Gökhan G.A., Bozova S. Trombospondin-1 expression correlates with angiogenesis in experimental cirrhosis. World J Gastroenterol 2008; 14 (14): 2213-2217.
- Eriksson K, Magnusson P, Dixelius J. et al. Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett 2003; 536 (1—3): 19—24.
- Jagavelu K, Routray C, Shergill U. et al. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the fiver. Hepatology 2010; 52 (2): 590-601.
- Melgar-Lesmes P, Pauta M., Reichenbach V. et al. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut 2011; 60 (10): 1404-1411.
- Huebert R.C., Jagavelu K, Hendrickson HI. et al. Aquaporin-1 promotes angiogenesis, fibrosis, and portal hypertension through mechanisms dependent on osmotically sensitive microRNAs. Am J Pathol 2011; 179 (4): 1851-1860.
- Sahin H, Borkham-Kamphorst E., Kuppe C. et al. Chemokine Cxcl9 attenuates fiver fibrosis-associated angiogenesis in mice. Hepatology 2012; 55 (5): 1610-1619.
- Staton С., Kumar I, Reed М., Brown N. Neuropilins in physiological and pathological angiogenesis. J Pathol 2007; 212 (3): 237-248.
- Fernandez M., Semela D., Brutx J. et al. Angiogenesis in liver disease. J Hepatol 2009; 50 (3): 604—620.
- Chaparro M., Sanz-Cameno P., Trapero-Marugan M. et al. Mechanisms of angiogenesis in chronic inflammatory liver disease. Ann Hepatol 2007; 6 (4): 208—213.
- Steib C.J. Kupffer cell activation and portal hypertension. Gut 2011; 60 (10): 1307-1308.
- Lochhead P.A., Gilley R., Cook S.J. ERK5 and its role in tumour development. Biochem Soc Trans 2012; 40 (1): 251—256.
- Dewhirst M.W., Cao Y., Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008; 8 (6): 425-437.
- КоM., Seo K.H., Han S.J. et al. Nuclear factor kappaB dependency of platelet-activating factor-induced angiogenesis. Cancer Res 2002; 62 (6): 1809-1814.
- Franceschini B., Ceva-Grimaldi G., Russo C. et al. The complex functions of mast cells in chronic human liver diseases. Dig Dis Sei 2006; 51 (12): 2248-2256.
- Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci 2002; 1 (7): 1899-1914.
- Coppie B.L., Bai S, Burgoon L.D., Moon JO. Hypoxia-inducible factor-la regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int 2011; 31 (2): 230-244.
- , CadoretA., Mourabit HE. etal. Origins and functions of fiver myofibroblasts. Biochim Biophys Acta 2013; 1832 (7): 948-954.
- Yokomori H, Oda M., Yoshimura K, Hibi T. Enhanced expressions of apelin on proliferative hepatic arterial capillaries in human cirrhotic fiver. Hepatol Res 2012; 42 (5): 508—514.
- Coulon S, Heindryckx E, Geerts A. et al. Angiogenesis in chronic fiver disease and its complications. Liver Int 2011; 31 (2): 146—162.
- Novo E., Povero D., Busletta C. et al. The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J Pathol 2012; 226 (4): 588—597.
- Novo E., Cannito S., Zamara E. et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol 2007; 170 (6): 1942—1953.
- Vanheule E., Geerts A.M., Van Huysse J. et al. An intravital microscopic study of the hepatic microcirculation in cirrhotic mice models: relationship between fibrosis and angiogenesis. Int J Exp Pathol 2008; 89 (6): 419-432.
- Kaur S, Tripathi D., Dongre K. et al. Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident fiver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J Hepatol 2012; 57 (6): 1193—1198.
- Chen C.H, Chang L.T, Tung W.C. et al. Levels and values of circulating endothelial progenitor cells, soluble angiogenic factors, and mononuclear cell apoptosis in fiver cirrhosis patients. J Biomed Sei 2012; 19: 66.
Related Articles
Юпатов Е.Ю.
Курманбаев Т.Е.
Тимошкова Ю.Л.