INTRODUCTION
Recent sequencing technologies can generate large volume of reads; an illumina HiSeq 2000 can generate 600 million pair-end reads of length 100 in 10 days. The high-throughput platform has been proven to answer various biological questions like mapping DNA-protein interactions and gene expression profiling. Mapping of the reads onto a reference genome is the first step, and a need for an extremely fast alignment tool for longer reads allowing three or more mismatches is a priority. Different tools have been designed for aligning short reads onto a reference genome, the most popular ones being MAQ (Li et al., 2008a), SOAP2 (Li et al., 2008b), Bowtie (Li et al., 2008a;b), and BWA (Langdon et al., 2015). The fastest existing central processing unit (CPU) based aligners can align 70 million read pairs to human genome with at most four mismatches which take >3.5 h and to align 1G read pairs, it takes >2 days to complete the alignment (Langdon et al., 2015). While graphics processing units (GPUs) are promising alternatives for increasing alignment speeds, one difficulty with GPU is that it works in a single-instruction multiple-thread (SIMT) mode, where the processors in the same unit must execute the same instruction and too many diverging branches in the execution path would force some of the processors to idle (Chi-Man et al., 2014). The introduced diverging branches cannot be determined until runtime. This issue is addressed by SOAP3, where hard patterns determine whether a pattern would introduce too many branches to stop the execution of hard patterns, group them and re-do the alignment of them in another round to reduce the idle time of processors. SOAP3 uses seed and hash look-up table algorithm to accelerate alignment, where both reads and the reference sequences are converted to numeric data type using 2-bits-per-base encoding. The value is then used as a suffix to check the look-up table to know how many bases are different. The algorithm outputs the identical alignments as that of dynamic programming and has been shown to run much faster (Li et al., 2008a; b).
Another important tool for alignment is BarraCUDA which works in four steps. The first step is to transfer the Burrows-Wheeler transform (BWT)-encoded reference sequence and sequence reads from disk to GPU using a 1-dimensional uint4 array to ensure fast data access. Sequence reads are loaded into GPU memory in batches and packed in a single continuous block to minimize internal fragmentations, and the data is bound to the texture cache to maximize the data throughput. The second step is CUDA thread assignments mapping a sequence read to a reference sequence is a data independent process and does not require any information from any of the other reads, so it employs data parallelism by assigning an alignment kernel thread to each of the individual sequencing reads and launching the GPU kernel with thousands of threads at the same time. The third step is the inexact sequence alignment using a depth-first search (DFS) GPU kernel using a backward search string-matching algorithm to look for alignments. The final step is a multiple kernel design, where the long sequence reads are divided into short fragments 32 base pairs and alignment is performed by multiple consecutive DFS kernel runs (Petr Klus et al., 2012).
CUSHAW aligner is designed based on the BWT and programmed using CUDA C++ parallel programming language and the performance evaluation of this aligner achieves significant speedups in terms of execution time and better alignment quality for paired-end alignments when compared with popular BWT-based aligners like Bowtie, BWA and SOAP2. The several important parameters like MMS (maximal number of mismatches allowed in the seed), MMR (maximal number of mismatches allowed in the full length of a read), QSS (maximal sum of quality scores at all mismatched positions in the seed), QSR (maximal number of quality scores at all mismatched positions in the full length of a read) and QSRB (maximal QSR among the currently selected best alignments, updated as the aligning process goes on) are explicitly declared (Yongchao et al., 2012).
SARUMAN (Semiglobal Alignment of short reads using CUDA and Needle MAN-Wunsch) is another important mapping approach that returns all possible alignment positions of a read in a reference sequence under a given error threshold. Alignments are computed in parallel on graphics hardware, facilitating a considerable speedup of this normally time-consuming step. They combine their filter algorithm with CUDA-accelerated alignments to improve alignment time. The tool is divided into two consecutive phases, mapping and aligning. Phase one consists of creating a qgram index and mapping the reads through qgrams, followed by phase 2 in which CUDA is used to compute the edit distance for candidate hits on the graphics card using a modified Needleman-Wunsch algorithm (Jochen et al., 2011).
This article focuses on compiling the aforementioned GPU based alignment tools on 70.7 million read pairs (Illumina HiSeq 2000) to align them on a human genome and check its efficiency (time sensitivity and alignment specificity) as compared to traditional CPU based alignment (Bowtie) tool. Resulting observations would help researchers choose the appropriate GPU alignment tool to suffice their computing needs.
MATERIALS AND METHODS
System requirements
The compilation of all the tools was conducted on a computer with a 3.07 GHz quad-core CPU and 24 G memory supported by a NVIDIA GTX 580 GPU card with 3G memory. Dataset has been chosen with 70.7 M read pairs, sequenced from YH1 Cell-line DNA using Illumina HiSeq 2000 (Wang et al., 2008). The datasets have read length of 100. All the tools were compiled with one GPU for maintaining consistent results.
Compilation codes
SOAP3: Building the 2BWT index with “./2bwt-builder human v36.1.fa”; convert the 2BWT index to the GPU2-BWT index with “./BGS-Build human v36.1.fa.index”; aligning with aligner with parameter -m for mismatches (from 0 to 3, default: 3) and -h for selecting all the alignments, applied as “./soap3_aligner human v36.1.fa.index QueryReads.fa 1000000 100 -m 2 -h 1”.
CUSHAW: Constructing BWT indices of genomes using “./bwt_index -a bwtsw human v36.1.fa” and aligning with “./cushaw human v36.1.fa.index -fasta QueryReads.fa -mms 3 –g 1”; with parameters –mms for mismatches (0-3) and GPU unit 1.
BarraCUDA: BWT-transformation of the human genome performed by “barracuda index human v36.1.fa”; followed by alignment with “barracuda aln human v36.1.fa QueryReads.fa > quicktest.sai”; followed by convertion of the SAI format to SAM by “barracuda samse quicktest.sai human v36.1.fa > quicktest.sam”
SARUMAN: Alignment performed by “./saruman-1.0.X-SM13 –r –g human v36.1.fa –e 3 –u 1” with parameters -u for GPU units (1) and number of mismatches –e (0-3).
RESULTS
GPU based alignment tools are extremely (5 to 37x times) faster than conventional CPU based alignment tool
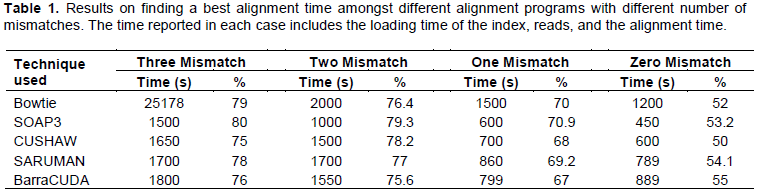
All the tested GPU based alignment tools are approximately 5 to 37x times significantly faster than CPU based alignment tool, Bowtie. The mismatches in alignment varied from zero to three mismatches, and in all of them alignment speed for GPU based alignment tools was faster. Where the time for Bowtie varied from 1200 to 25178 s, the timings for GPU alignment tools varied from 450 to 1700 s amongst different mismatches. The comparison of these alignment timings are shown in Table 1. It was predicted that the GPU based alignment tools usage should replace all the CPU based alignment tools in future for faster’s alignment results.
SOAP3 outperforms most of the compared GPU and CPU based alignment tools with respect to alignment times (sensitivity)
As shown in Figure 1, amongst compared GPU based alignment tools, SOAP3 exceeds the alignment timing speed. The timing varied from 450 to 1500 s. This was followed with CUSHAW, SARUMAN and BarraCUDA at timings 600 to 1650, 789 to 1700 and 799 to 1800 s, respectively. With larger reference genomes (humans, mice, mammals), the use of SOAP3 amongst other GPU based alignment tools is recommended.
No significant differences are seen with the specificity (alignment coverage) for either CPU or GPU based alignment tools
Although, significant differences in alignment timings amongst GPU based alignment tools was observed, not much difference in coverage area was seen (Figure 2). The coverage varied from 50 to 80% for SOAP3 including Bowtie (amongst all the mismatches). More coverage is important during alignment process, and presently it is an important research topic amongst different groups in the field of sequencing.
DISCUSSION
When aligning with up to three mismatches, the HiSeq 2000 dataset revealed that SOAP3 is at least 5 to 37x times faster than other GPU based alignment tools and Bowtie is the slowest in this setting. SOAP3 favors large dataset as it takes longer time to load the index. Bowtie is a heuristics based tool, while SOAP3 reports all the alignments, and it aligns slightly more reads than Bowtie (Table 1). For computers with multiple CUDA-capable GPUs, BarraCUDA automatically selects the best GPU based on number of stream processors and the amount of graphics memory available to the software. Users can specify which CUDA device software is to be executed (-C parameter), this can make BarraCUDA slightly more efficient than SOAP3 but not optimal. It has been shown that the multiple GPUs show a better scalability than CPUs. An alignment throughput of BarraCUDA with 1 Tesla M2050 GPU is similar to that of BWA with 6 CPU cores (Xeon X5670 2.93 GHz with 8 GB DDR3 RAM). Furthermore, just by using BarraCUDA with two GPUs can outperformed BWA using all 12 cores (2 × Xeon X5670s) at 2.5 Mbp/s (Petr Klus et al., 2012). Another proposed method to accelerate the process is to combine the CUDA alignment module with filter algorithms on graphics adapter, which can reduce the memory usages. Another planned development is a native support for color space data as generated by the SOLiD sequencing system. Given the present observations, it was predicted that the GPU based alignment tool usage will replace all the CPU based alignment tools in the future and currently SOAP3 comes out to be the fastest alignment tool for aligning up to and not limited to 70.7 M read pairs.
The authors have not declared any conflict of interests.
Support from the Georgia State University Information Technology Department (GSU IT) for server space is gratefully acknowledged.
REFERENCES
Chi-Man L, Ruibang L, Tak-Wah L (2014). GPU-Accelerated BWT Construction for Large Collection of Short Reads. CiteSeer. 2014.
|
|
Jochen B, Tobias J, Daniel D, Sebastian J, Jörn K, Jens S, Alexander G (2011). Exact and complete short-read alignment to microbial genomes using Graphics Processing Unit programming. Bioinformatics 27(10):1351-1358.
Crossref
|
|
|
Li H (2008a). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research 18:1851-1858.
Crossref
|
|
|
Li R (2008b). SOAP: short oligonucleotide alignment program. Bioinformatics 24:713-714.
Crossref
|
|
|
Langdon WB, Lam BY, Petke J, Harman M (2015). Improving CUDA DNA Analysis Software with Genetic Programming. Proceedings of the 2015 Annual Conference on Genetic and Evolutionary Computation - GECCO '15.
Crossref
|
|
|
Petr Klus K, Simon L, Dag L, Ming SC, Graham P, Ian M, Giles SHY, Brian YHL (2012). BarraCUDA - a fast short read sequence aligner using graphics processing units. BMC Research Notes 5:27.
Crossref
|
|
|
Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y (2008). The diploid genome sequence of an Asian individual. Nature 456(7218):60.
Crossref
|
|
|
Yongchao L, Bertil S, Douglas LM (2012). CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows–Wheeler transform. Bioinformatics 28(14):1830-1837.
Crossref
|